

Abstract
Early relapse detection in acute myeloid leukemia is possible using standardized real-time quantitative polymerase chain reaction (RQ-PCR) protocols. However, optimal sampling intervals have not been defined and are likely to vary according to the underlying molecular lesion. In 74 patients experiencing hematologic relapse and harboring aberrations amenable to RQ-PCR (mutated NPM1 [designated NPM1c], PML-RARA, RUNX1-RUNX1T1, and CBFB-MYH11), we observed strikingly different relapse kinetics. The median doubling time of the CBFB-MYH11 leukemic clone was significantly longer (36 days) than that of clones harboring other markers (RUNX1-RUNX1T1, 14 days; PML-RARA, 12 days; and NPM1c, 11 days; P < .001). Furthermore, we used a mathematical model to determine frequency of relapse detection and median time from detection of minimal residual disease to hematologic relapse as a function of sampling interval length. For example, to obtain a relapse detection fraction of 90% and a median time of 60 days, blood sampling every sixth month should be performed for CBFB-MYH11 leukemias. By contrast, in NPM1c+/FLT3-ITD−, NPM1c+/FLT3-ITD+, RUNX1-RUNX1T1, and PML-RARA leukemias, bone marrow sampling is necessary every sixth, fourth, and fourth and second month, respectively. These data carry important implications for the development of optimal RQ-PCR monitoring schedules suitable for evaluation of minimal residual disease–directed therapies in future clinical trials.
Introduction
Relapse remains the event that heralds ultimate treatment failure for most acute myeloid leukemia (AML) patients.1 Thus, whereas reacquisition of complete remission (CR) is often possible, it always poses a greater therapeutical challenge than the initial cytoreduction, most probably because of selection for therapy-resistant clones.1,2
Consequently, detection of impending relapse remains a major challenge in these patients. Several tools with various sensitivities are available for this task, most notably multicolor flow cytometry (sensitivity as low as 0.01%)3 and real-time quantitative polymerase chain reaction (RQ-PCR; sensitivity as low as 0.0001%).4,5 RQ-PCR assays are being applied to fusion transcripts, such as PML-RARA,6-11 CBFB-MYH11,8,10,12-14 RUNX1-RUNX1T1,8,10,15-19 and DEK-CAN;20-22 overexpressed genes, such as WT123-26 and PRAME;27 or mutated genes, such as NPM1.28-32 There is evidence to suggest that assay sensitivity not only varies between minimal residual disease (MRD) markers, but can also differ significantly between patients possessing the same molecular target.9,10,32 Furthermore, as follow-up MRD samples are often collected sparsely, even in prospective studies, it has hitherto been difficult to provide firm recommendations for sampling, not only regarding intervals, but equally so with regard to source of sample material (ie, bone marrow [BM] or peripheral blood [PB]).
To address these issues, we recently used the Wilms tumor gene 1 (WT1) as a molecular marker to derive a mathematical model enabling the delineation of several quantitative parameters related to relapse kinetics, such as the power to detect molecular relapse (MR; relapse detection fraction [RDF]) and the median time (tm) from molecular positivity to hematologic relapse (HR) for different sampling intervals.33
Given that the sensitivity of the WT1 assay is inferior to assays detecting recurrent molecular aberrations such as fusion transcripts and mutations, we in this study present data collected from 3 centers handling large numbers of MRD samples. By analyzing relapsing patients with several prerelapse samples available and applying our mathematical model, we have now been able to delineate relapse kinetics in patients harboring 4 different target transcripts, namely RUNX1-RUNX1T1, CBFB-MYH11, PML-RARA, and mutated NPM1 (designated NPM1c34 ). Application of the model, which takes into account differences in assay sensitivity and relapse kinetics, will allow development of monitoring schedules tailored according to the molecular lesion, suitable for evaluating the clinical utility of MRD-directed therapies in multicenter clinical trials.
Methods
Patient samples
Patient samples were analyzed at the Laboratory of Immunohematology (IHL), Aarhus University Hospital, from October 1995 to November 2007; the Munich Leukemia Laboratory from July 2005 to March 2008; and the Department of Medical and Molecular Genetics/Molecular Oncology Diagnostics Unit, Guy's Hospital from May 2002 to March 2009. All patients were diagnosed and followed for MRD detection using standard published methods.8-10,32,35 MRD analyses were conducted with informed patient consent in accordance with the Declaration of Helsinki and subject to ethics committee approval from all participating institutions. The patients with PML-RARA acute promyelocytic leukemia were treated with all-trans retinoic acid and anthracycline-based chemotherapy, as detailed in Grimwade et al.9 Non–acute promyelocytic leukemia (APL) patients were treated with standard combination therapy regimens, as described.10,32,33,36
MRD determination and reporting
MRD values were calculated using the ΔΔ cycle threshold (Ct) or the absolute quantification method using plasmid standards (Ipsogen), as described by Beillard et al,37 according to the laboratory and molecular target (for NPM1c quantification, see Schnittger et al32 ). Samples with low-quality RNA (defined as a threshold cycle number of the control gene ß2-microglobulin exceeding 25 and/or a threshold cycle number of the control gene Abelson exceeding 30) were excluded.
Assays were run in duplicate (NPM1c) or triplicate (other markers), and in accordance with Europe against Cancer criteria, amplification in at least 2 of 3 replicates with Ct values up to 40 (threshold 0.1 [0.05 for PML-RARA]) was required to define a result as polymerase chain reaction (PCR) positive for the MRD marker in question.37 For the depiction of relative values on an absolute scale, data were transformed from ΔCt values using the PCR efficiencies found in routine laboratory assay testing.
Definition of MR, HR, and molecular CR
Definitions of MR, HR, and molecular CR (mCR) followed the recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia,38 with the modifications described in “Low-level positive expression in patients in continuous CR.” Thus, patients were considered having a MR at first recurrence of MRD marker PCR positivity, except for those followed using the NPM1c or RUNX1-RUNX1T1 molecular markers in which a MRD level above 5 × 10−5 or 1 × 10−4, respectively, was required to define MR. Similarly, patients with the 2 latter markers were considered in mCR if PCR expression levels dropped to below these thresholds.
Inclusion and exclusion criteria
To be eligible for relapse kinetics analysis, patients needed to enter CR, have PCR samples taken after the discontinuation of chemotherapy, but before HR, and not receive pre-emptive treatment upon MR. Thus, patients PCR positive after treatment, who were not given further high-dose chemotherapy, could be included in this part of the study (n = 5).
To be eligible for relapse modeling based on PCR conversion, patients needed to enter mCR, have samples taken the last year before HR, and not receive pre-emptive treatment upon MR. Thus, patients experiencing a HR with no previous positive samples could be included (n = 28). One patient with a central nervous system relapse was excluded from both analyses.
Statistical analyses
To compare relative levels of expression of the molecular target at diagnosis and at relapse, as well as increments in normalized leukemic transcripts before HR, Wilcoxon rank-sum test was used. To compare the performance of PB versus BM testing, the number of paired MRD measurements in which only the BM was positive was compared with the number of MRD measurements in which only the PB was positive using the binomial distribution. These analyses were restricted to paired samples in which the BM and PB yielded RNA of comparable quality, as indicated by the respective levels of control gene expression.
Modeling of relapse patterns
Relapse patterns were modeled as described in Ommen et al.33 Briefly, prerelapse samples were divided into monthly intervals based on how long before time of HR they were taken. Patients were considered RQ-PCR negative or positive in all intervals between 2 negative or positive samples, respectively. The fraction of positive samples in each interval was then plotted against time to relapse. For a given positive sampling interval, I, the chance of detecting the relapse, RDF, was then given by:

where F(t) describes the distribution of the time span from when the leukemic burden exceeds the sensitivity of a given MRD marker to HR. This function can be approximated based on the distribution of individual patients' conversion to PCR positivity before HR, as described in Ommen et al.33 The median time from MR to HR, tm, can be found by solving the integral equation:
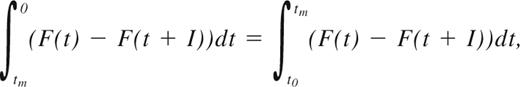
where t0 is the intercept with the x-axis.33 In this manner, RDFs and tm for sampling intervals 1, 2, 3, 4, 6, 9, and 12 months were found for each molecular marker in both PB and BM. F(t) is approximated using high-grade polynomials for each of the MRD markers. The polynomials used can be found in supplemental Document 1 (available on the Blood website; see the Supplemental Materials link at the top of the online article).
Results
MRD marker expression at diagnosis and relapse
A major determinant of the capacity for RQ-PCR to detect residual AML in a background of normal hematopoietic cells is assay sensitivity, which depends on the relative level of expression of the MRD target in leukemic blasts (reviewed in Freeman et al5 ). To compare assay sensitivities, we determined the MRD marker expression in relation to control gene expression in BM at diagnosis from 365 patients (151 PML-RARA, 31 CBFB-MYH11, 42 RUNX1-RUNX1T1, and 141 NPM1c) and at HR for all relapsing patients.
As seen from supplemental Figure 1 and supplemental Table 1, median expression of NPM1c was higher than that of the fusion transcript MRD markers that exhibited comparable levels of expression. Whereas diagnosis levels were generally higher than relapse levels, median values were within the same order of magnitude, allowing the use of diagnosis expression level as a measurement for MRD assay sensitivity.
Low-level positive expression in patients in continuous CR
Simple qualitative PCR positivity has been reported in several cases in patients who did not later relapse.39-41 To investigate this situation in this large cohort of patients, we analyzed the number of positive samplings in patients who had earlier tested negative (for PML-RARA patients, 2 consecutive negative samples were required, as described in Grimwade et al9 ). For all 4 aberrations, a small percentage of positive samples could in fact be observed, the highest fraction in NPM1c patients (26% of continuous CR [CCR] samplings), possibly due to the higher level of MRD marker transcripts in these cells or in some cases due to cross-reactivity of the mutant assay with the wild-type allele.10 However, when a cutoff of 5 × 10−5, relative to the diagnostic level, was used to define MR, only 1 positive reaction was not followed by a HR (of a total of 104 CCR determinations). Applying a similar cutoff level for RUNX1-RUNX1T1 patients at 1 × 10−4 resulted in 1 positive reaction of 87 CCR determinations not followed by a HR. By contrast, positive samplings in CCR using the CBFB-MYH11 molecular marker occurred in only 3 samples, accounting for 4.8% of the CCR samples. For this aberration, we therefore chose not to use a MR cutoff level, and resorted to the requirement of 2 consecutive positive samplings to define a MR given that CBFB-MYH11 relapses are generally indolent, allowing for such an approach.42 For PML-RARA follow-up, positive samples were rare, and we chose not to use a cutoff threshold in the case of this aberration. For the 2 assays (NPM1c and RUNX1-RUNX1T1) in which thresholds were introduced to exclude very low-level positivity not followed by a HR, the maximum sensitivity corresponds to the level of the threshold. As the sensitivity of the assays using the other 2 MRD markers is approximately 1 × 10−4 (with a range of 1 × 10−5 to 1 × 10−3 reflecting differences in the relative level of expression of the MRD target in the leukemic blasts between patients, as well as variation in the quality of follow-up samples), we found that the sensitivities of MRD detection, and thereby the ability to detect MR, using the 4 different markers are comparable (Figure 1).

Low-level positive RQ-PCR samplings in patients in CCR. x-axis, MRD target; y-axis, MRD marker expression level relative to diagnostic level, arbitrarily set to 1. Numbers below the figure depict the number of low-level positive samples, and negative samples in patients with available diagnostic levels. Suggested cutoff levels for RUNX1-RUNX1T1 and NPM1c are shown in red bars and given in numbers. Sensitivity of each test can be understood as the lower of the difference between diagnostic level and the sample quality-dependent sensitivity (typically between 1 × 10−5 and 1 × 10−3) and cutoff level (NPM1c and RUNX1-RUNX1T1).
Low-level positive RQ-PCR samplings in patients in CCR. x-axis, MRD target; y-axis, MRD marker expression level relative to diagnostic level, arbitrarily set to 1. Numbers below the figure depict the number of low-level positive samples, and negative samples in patients with available diagnostic levels. Suggested cutoff levels for RUNX1-RUNX1T1 and NPM1c are shown in red bars and given in numbers. Sensitivity of each test can be understood as the lower of the difference between diagnostic level and the sample quality-dependent sensitivity (typically between 1 × 10−5 and 1 × 10−3) and cutoff level (NPM1c and RUNX1-RUNX1T1).
Relapse kinetics
The advantage of using the RQ-PCR methodology in MRD follow-up is its potential for revealing relapse kinetics in the individual patient, provided that enough RQ-PCR–positive samples have been obtained before HR (Table 1).
Study subjects
| MRD marker | NPM1c+ | PML-RARA | CBFB-MYH11 | RUNX1-RUNX1T1 |
|---|---|---|---|---|
| No. of patients | 180 | 406* | 47 | 56 |
| Patients with HR | 54 | 22* | 19 | 20 |
| Patients in CCR | 126 | 363* | 28 | 36 |
| CCR samples | 193 | 1865 | 151 | 170 |
| Relapse kinetics | ||||
| Patients included | 22 | 8 | 9 | 7 |
| Patients excluded | 32 | 13 | 10 | 13 |
| Reasons for exclusion: | ||||
| No MR/HR sample | 32 | 9 | 10 | 12 |
| Administration of pre- emptive therapy in MR | 0 | 4 | 0 | 1 |
| CNS relapse | 0 | 1 | 0 | 0 |
| Relapse modeling | ||||
| Patients included | 31 | 13 | 14 | 11 |
| Samples | 70 | 61 | 59 | 46 |
| Patients excluded | 23 | 8 | 5 | 9 |
| Reasons for exclusion: | ||||
| No mCR | 3 | 1 | 0 | 1 |
| No CR samples† | 20 | 4 | 5 | 7 |
| Administration of pre- emptive therapy in MR | 0 | 3 | 0 | 1 |
| CNS relapse | 0 | 1 | 0 | 0 |
| MRD marker | NPM1c+ | PML-RARA | CBFB-MYH11 | RUNX1-RUNX1T1 |
|---|---|---|---|---|
| No. of patients | 180 | 406* | 47 | 56 |
| Patients with HR | 54 | 22* | 19 | 20 |
| Patients in CCR | 126 | 363* | 28 | 36 |
| CCR samples | 193 | 1865 | 151 | 170 |
| Relapse kinetics | ||||
| Patients included | 22 | 8 | 9 | 7 |
| Patients excluded | 32 | 13 | 10 | 13 |
| Reasons for exclusion: | ||||
| No MR/HR sample | 32 | 9 | 10 | 12 |
| Administration of pre- emptive therapy in MR | 0 | 4 | 0 | 1 |
| CNS relapse | 0 | 1 | 0 | 0 |
| Relapse modeling | ||||
| Patients included | 31 | 13 | 14 | 11 |
| Samples | 70 | 61 | 59 | 46 |
| Patients excluded | 23 | 8 | 5 | 9 |
| Reasons for exclusion: | ||||
| No mCR | 3 | 1 | 0 | 1 |
| No CR samples† | 20 | 4 | 5 | 7 |
| Administration of pre- emptive therapy in MR | 0 | 3 | 0 | 1 |
| CNS relapse | 0 | 1 | 0 | 0 |
MRD indicates minimal residual disease; HR, hematologic relapse; CR, complete remission; CCR, continuous CR; MR, molecular relapse; mCR, molecular CR; and CNS, central nervous system.
Numbers do not add, as some patients were successfully treated in first MR.
In the 12 months prior to HR, there were no CR samples.
In Figure 2 the CBFB-MYH11 patients display a slower rate of rise of leukemic transcripts before HR than all the other aberrations (median BM doubling time 36 days, range 7.4-175 days, versus all other aberrations; P < .001). Compared with this, the incremental rise in NPM1c transcripts was more rapid (median BM doubling time 11 days, range 2.2-33 days; P = .002); however, there was marked heterogeneity with some patients experiencing indolent relapses, whereas others exhibited the shortest doubling times in the dataset. A major reason for this duality was found to be the presence or absence of the FLT3-ITD aberration (NPM1c+/FLT3-ITD−, median BM doubling time 15 days, range 2.2-33 days; NPM1c+/FLT3-ITD+, median BM doubling time 7.4 days, range 3.0-14 days; P = .031). RUNX1-RUNX1T1 relapsing clone reappearance was faster than CBFB-MYH11 growth (median BM doubling time 14 days, range 12-51 days; P = .105), as was PML-RARA clone growth (median BM doubling time 12 days, range 4.2-462 days; P = .093), with the exception of 1 patient who displayed the slowest relapse growth rate in the entire cohort (BM and PB doubling times of 462 and 54 days, respectively). Both these leukemias progressed with approximately the same speed as the NPM1+/FLT3-ITD− ones (P = .97 and .93, respectively), but slower than the NPM1+/FLT3-ITD+ leukemias (P = .02 and .05, respectively).

Relapse kinetics according to molecular lesion. Increasing MRD marker levels are shown in patients between time of molecular relapse and diagnosis of HR. Expression normalized to HR level (set to 1). x-axis, Time before HR. y-axis, Normalized MRD levels with PB and BM values depicted separately.
Relapse kinetics according to molecular lesion. Increasing MRD marker levels are shown in patients between time of molecular relapse and diagnosis of HR. Expression normalized to HR level (set to 1). x-axis, Time before HR. y-axis, Normalized MRD levels with PB and BM values depicted separately.
Relapse modeling based on conversion to PCR positivity
In some patients subjected to longitudinal RQ-PCR testing, no positive samples before HR will be obtained, either because of very rapid clone growth, poor sampling quality giving rise to false negative results, or simply unfortunate scheduling of MRD assessment relative to the timing of recurrent PCR positivity. We devised a mathematical model, which takes this lack of data into account and allows inclusion of information from such patients.33 As can be seen in Figure 3, which depicts time before relapse as a function of the fraction of positive samples in each interval for each molecular marker, we were able to confirm and extend the relapse kinetics findings shown in Figure 2. Thus, CBFB-MYH11 emerged as the MRD marker with the longest lag from MR to HR (50% of the patients tested RQ-PCR positive in BM 8 months before relapse; Figure 3C). By comparison, RUNX1-RUNX1T1 relapses showed more rapid relapse kinetics with 50% being positive in BM as close as 3 months before relapse (Figure 3D). Moreover, the dichotomy observed for NPM1c transcript-based relapse detection was preserved in the model with a 3-month difference in when 50% of FLT3-ITD− and FLT3-ITD+ patients became PCR positive (6.5 months vs 3.5 months before relapse; Figure 3A). For PML-RARA–positive relapses, the heterogeneity was even more pronounced, with 1 relapse being detectable 14 months before HR, and 2 being undetectable 71 days and 41 days before HR, respectively (50% detectable 3.5 months before HR; Figure 3B).

Conversion to PCR positivity. x-axis, Time, HR occurred at t = 0. y-axis, Fraction-positive samples in each prerelapse interval. Median number of evaluable MRD profiles in each interval is shown for each molecular marker (panels A-D; range in parentheses). Solid lines, BM; broken lines, PB.A: NPM1c: blue, FLT3-ITD−; red, FLT3-ITD+. B: PML-RARA. C: CBFB-MYH11. D: RUNX1-RUNX1T1.
Conversion to PCR positivity. x-axis, Time, HR occurred at t = 0. y-axis, Fraction-positive samples in each prerelapse interval. Median number of evaluable MRD profiles in each interval is shown for each molecular marker (panels A-D; range in parentheses). Solid lines, BM; broken lines, PB.A: NPM1c: blue, FLT3-ITD−; red, FLT3-ITD+. B: PML-RARA. C: CBFB-MYH11. D: RUNX1-RUNX1T1.
A recurring question when RQ-PCR is used for MRD detection has been to what extent PB can substitute for BM sampling. Comparing PB versus BM-based MR detection, it is now apparent that for the majority of PML-RARA cases, BM sampling is superior. Thus, in 7 paired samplings, MR was detected in BM only in 5 cases and in PB only in none (P = .031). In Figure 3C and D, in contrast suggests that PB and BM sampling are equally useful for CBFB-MYH11 and RUNX1-RUNX1T1, even though the number of paired samplings was too low to draw any firm conclusions.
Taking advantage of the mathematical modeling, we were finally able to calculate the RDFs and tm to hematologic relapse as a function of sampling interval using the formulas described in “Modeling of relapse kinetics” (Figure 4). For NPM1c-based follow-up, modeling was done for the subgroups containing and lacking FLT3-ITD separately. As conclusions regarding NPM1c-based PB sampling would be based on very few patients (median number of MRD courses per interval 2.5), no modeling was done for NPM1c-based follow-up in this tissue. For comparative purposes and considering that some patients are negative for all the markers described in this study, we included WT1 data that complement the ones presented in this study, resulting in more than 80% of AML patients having a valid molecular marker for MRD detection.25

Comparisons between different molecular markers regarding RDF and tm. x-axis, Sampling time in months. y-axis, RDF (top), median time from MR to HR (tm; bottom). Horizontal black lines, top panels: RDF equaling 90%. Bottom panels, tm equaling 60 days. Blue lines in top panel represent sampling interval necessary to ensure 90% relapse detection. Blue lines in bottom panels show corresponding tm values. Solid lines, BM; broken lines, PB; NPM1c-based MRD follow-up: black line, FLT3-ITD− and red line, FLT3-ITD+. PML-RARA–based MRD follow-up: PB sampling intervals above 6 months not shown as model handles situation of long sampling intervals in very heterogeneous populations poorly.
Comparisons between different molecular markers regarding RDF and tm. x-axis, Sampling time in months. y-axis, RDF (top), median time from MR to HR (tm; bottom). Horizontal black lines, top panels: RDF equaling 90%. Bottom panels, tm equaling 60 days. Blue lines in top panel represent sampling interval necessary to ensure 90% relapse detection. Blue lines in bottom panels show corresponding tm values. Solid lines, BM; broken lines, PB; NPM1c-based MRD follow-up: black line, FLT3-ITD− and red line, FLT3-ITD+. PML-RARA–based MRD follow-up: PB sampling intervals above 6 months not shown as model handles situation of long sampling intervals in very heterogeneous populations poorly.
Once more, CBFB-MYH11 leukemia displayed the most tardy relapses with high RDFs and tms for sampling intervals as long as 6 months (PB, RDF 90% and tm 180 days; BM, RDF 85% and tm150 days). Sampling interval of this length also yielded satisfactory results in the group of patients with NPM1c leukemia without FLT3-ITD (6-month intervals, BM: RDF 90%, tm 120 days). As an intermediate group, for NPM1c (with FLT3-ITD) and RUNX1-RUNX1TI leukemias, application of a 3- to 4-month sampling frequency still yielded satisfactory relapse detection (4-month intervals, NPM1c+FLT3-ITD+, BM, RDF 85% and tm 65 days; RUNX1-RUNX1TI PB, RDF 75% and tm 55 days; BM, RDF 95% and tm 85 days). Somewhat surprisingly, relapse detection by WT1 fell into this category too, at least when BM was used (4-month intervals, WT1; BM, RDF 95% and tm 75 days). Due to the occurrence of 2 patients who were negative in BM close to HR, PML-RARA relapses were the most difficult to detect, and 2-month BM sampling will be necessary to obtain the relapse detection efficiencies of the other markers (2-month intervals, PML-RARA; RDF 95%, tm 70 days).
Discussion
Close molecular monitoring using RQ-PCR in AML patients in CR holds the promise of detecting subclinical levels of residual disease in time to institute treatment intervention to prevent overt relapse. Despite an impressive body of data showing that RQ-PCR is excellently suited for early detection of AML relapses,6-28,30-32 few investigators have taken clinical action on these findings, although it is by now evident that the risk of a false positive result of a RQ-PCR is minimal, at least when the thresholds to exclude irrelevant low-level amplification in CCR are used, and especially so when molecular conversion is confirmed in a subsequent sample.9,33,43,44 To date, benefit from early salvage after conversion has been shown in PML-RARA APL,9,45,46 and in a preliminary report on the use of donor-lymphocyte infusion upon recurrent WT1 positivity after allogeneic transplantation,47 but has not been otherwise evaluated in non-APL AML patients.
Major reasons for this lack of translation of a powerful and technically standardized molecular method into clinical decision making beyond APL include uncertainty as to (1) the most informative schedules for MRD monitoring, (2) the most appropriate management of confirmed molecular relapse, and (3) whether early treatment intervention is likely to confer any clinical benefit compared with retreatment in overt relapse. To develop optimal MRD monitoring schedules to allow reliable assessment of the clinical utility of MRD monitoring in non-APL patients within large scale clinical trials, we have analyzed data from a very large cohort of patients subject to hematologic relapse (n = 114).
We used 2 different ways of describing the behavior of the leukemic clone before HR. First, we used the quantitative data obtained from patients in whom MR was identified to describe relapsing clone growth before HR. Assuming a constant doubling time, we were able to compare the different leukemia subsets and show that CBFB-MYH11 leukemia displayed slower leukemic clone growth than AML with mutant NPM1 or the RUNX1-RUNX1T1 fusion and, with a single exception, PML-RARA+ APL. Interestingly, but perhaps not surprisingly, NPM1c+/FLT3-ITD+ AML displayed relapses occurring significantly faster than all other molecular subtypes studied.
In the other model presented in this study, we examined pre-HR conversion to PCR positivity. This approach has the advantage of allowing the inclusion of information on patients for whom MR was not necessarily detected before HR. In addition, the assumption of a constant doubling time of the malignant clone is not necessary, although the cost is the lower degree of integration of quantitative values in this model. Thus, the 2 models complement each other, and it is reassuring that the results using these distinct approaches were near identical.
By directly comparing paired samples of PB and BM, we were able to show that PML-RARA BM MRD testing is superior to that of PB. Moreover, in the absence of a suitable number of paired samples for the other aberrations, the PCR positivity conversion model suggests that PB and BM are comparable for MRD detection in core binding factor leukemia. This is an encouraging conclusion, as PB sampling is much less troublesome for the patients, even if slightly higher sampling frequencies are necessary. However, further analyses are needed, especially regarding NPM1c-based MRD detection in PB, as our data do not support any definitive conclusions in this matter.
One great challenge when optimizing MRD follow-up is to determine the optimal sampling interval. The model presented in this study allows for the evaluation of suitable sampling intervals for the different analyzed markers.
Establishing guidelines for follow-up sampling should include considerations about their predictive value of MRD assessment, the options for intervention, as well as cost-benefit estimations. Thus, PB sampling every 6 months in patients with CBFB-MYH11 leukemia from PB will result in a RDF of 90% and a tm of 180 days. Even with this sampling cadenza, only 10% of relapses will be missed before hematologic relapse. Moreover, if the first sample is obtained 3 months after discontinuation of therapy and each patient is followed for only 3 years (given the literature on AML relapse48 and that 15 of 16 CBFB-MYH11 relapses in the study cohort occurred during the first 3 years after diagnosis), based on the present study, only 14 MRD samples would have to be taken to detect 1 MR. Given such considerations, which can be applied equally well to other RQ-PCR assays, it will be apparent that molecular monitoring might prove to be cost effective with its promise of early, possibly less intensive, intervention.
In conclusion, we in this study present data enabling us to model relapse kinetics in 4 common AML subtypes. We show great difference between these subtypes, and show that the studied markers are generally superior to nonleukemia-specific marker WT1. Furthermore, we show that this superiority can be used in the core binding factor leukemias to use PB as the sampling tissue. These data should be useful in cost-benefit calculations regarding MRD monitoring implementation, in clinical decision making in the individual patient, and in statistical power calculations when designing clinical trials.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Drs Morten Frydenberg and Steffen Hokland for statistical help. We thank Dr Robert Hills for helpful discussions, and D.G. gratefully acknowledges Prof Alan Burnett and the United Kingdom National Cancer Research Institute AML Working Group for access to data from APL patients treated in the Medical Research Council AML15 trial. P.H. thanks Prof Finn Bo Petersen, Intermountain Health Care Center, Salt Lake City, for helpful discussions.
This study was supported by grants from the Danish Medical Research Council, the Danish Cancer Society, the John and Birthe Meyer Foundation, and the Karen Elise Jensen Foundation (P.H.). D.G. and J.V.J. gratefully acknowledge support from the Leukemia Research Fund (Great Britain) and the European LeukemiaNet. This study was conducted within the framework of the MRD Workpackage (WP12) of the European LeukemiaNet.
Authorship
Contribution: H.B.O. and P.H. designed the study, analyzed the data, and wrote the first draft of the manuscript; S.S. was responsible for the analyses performed at Munich Leukemia Laboratory; J.V.J. and D.G. were responsible for the analyses performed at the Department of Medical and Molecular Genetics; I.B.O. analyzed the data; H.H. was responsible for the clinical care of the pediatric patients followed at the IHL; and M.Ø. was responsible for the analyses performed at IHL. All authors made significant contributions to the final manuscript.
Conflict-of-interest disclosure: S.S. in part owns Munich Leukemia Laboratory. The remaining authors declare no competing financial interests.
Correspondence: Peter Hokland, Aarhus University Hospital, Tage-Hansens Gade 2, Århus, Denmark 8000; e-mail: phokland@ki.au.dk.
