

KIT receptor tyrosine kinase mutations are implicated as a prognostic factor in adults with core binding factor (CBF) acute myeloid leukemia (AML). However, their prevalence and prognostic significance in pediatric CBF AML is not well established. We performed KIT mutational analysis (exon 8 and exon 17) on diagnostic specimens from 203 pediatric patients with CBF AML enrolled on 4 pediatric AML protocols. KIT mutations were detected in 38 (19%) of 203 (95% CI, 14%-25%) patient samples of which 20 (52.5%) of 38 (95% CI, 36%-69%) involved exon 8, 17 (45%) of 38 (95% CI, 29%-62%) involved exon 17, and 1 (2.5%; 95% CI, 0%-14%) involved both locations. Patients with KIT mutations had a 5-year event-free survival of 55% (± 17%) compared with 59% (± 9%) for patients with wild-type KIT (P = .86). Rates of complete remission, overall survival, disease-free survival, or relapse were not significantly different for patients with or without KIT mutations. Location of the KIT mutation and analysis by cytogenetic subtype [t(8;21) vs inv(16)] also lacked prognostic significance. Our study shows that KIT mutations lack prognostic significance in a large series of pediatric patients with CBF AML. This finding, which differs from adult series and a previously published pediatric study, may reflect variations in therapeutic approaches and/or biologic heterogeneity within CBF AML. Two of 4 studies included in this analysis are registered at http://clinicaltrials.gov as NCT00002798 (CCG-2961) and NCT00070174 (COG AAML03P1).
Introduction
Core binding factor (CBF) acute myeloid leukemia (AML) is characterized by the presence of a t(8;21) (q22;q22) or inv (16) (p13.1q22)/t(16;16) (p13.1;q22) [hereafter referred to as inv(16)] chromosomal rearrangement and is observed in approximately 20% to 30% of pediatric AML cases.1 Although overall survival (OS) for pediatric patients with CBF AML is superior to that of pediatric patients with AML with normal cytogenetics, a subset of these patients do quite poorly, suggesting that disease characteristics of this population are not as homogeneous as their cytogenetic definition and that additional mutational events may affect disease response.1,–3 KIT is a proto-oncogene located on chromosome band 4q11-12 and encodes a 145-kDa transmembrane glycoprotein that is a member of the type III receptor tyrosine kinase family.2,4,5 Stem cell factor promotes KIT dimerization and transphosphorylation when bound, thereby activating downstream signaling pathways integral to proliferation, differentiation, and survival of hematopoietic stem cells.5 Ligand independent activation of KIT results from mutations in the extracellular portion of the receptor (exon 8), transmembrane and juxtamembrane domains (exons 10 and 11, respectively), and activation loop of the tyrosine kinase domain (exon 17).5 Mutations of KIT are overall infrequent in adult AML (2%-8%) and tend to cluster within the activation loop (exon 17) and a region of the extracellular domain integral to receptor dimerization (exon 8).5 Their prevalence is much higher, however, in adult patients with CBF AML (6%-48%)2,6,,,,–11 and may be associated with worse clinical outcome in this patient population.2,6,9,–11 The incidence and clinical significance of KIT mutations in pediatric CBF AML is less clear because studies have been limited to a small number of patient samples.12,,–15
We screened a total of 203 diagnostic samples obtained from pediatric patients with CBF AML enrolled on Pediatric Oncology Group (POG) protocol 9421, Children's Cancer Group (CCG) protocols 2891 and 2961 and Children's Oncology Group (COG) protocol AAML03P1 for evidence of mutations in KIT exon 8 or 17. Our study, which represents the largest pediatric analysis to date, provides insight into the incidence and prognostic implications of KIT mutations in pediatric patients with de novo CBF AML treated on serial pediatric cooperative clinical trials.
Methods
Pediatric patients with previously identified CBF AML and enrollment on pediatric cooperative trials POG-9421, CCG-2891, CCG-2961, or COG protocol AAML03P1 were eligible for this study. This study was approved by the Fred Hutchinson Cancer Research Center Institutional Review Board and the COG Myeloid Disease Biology Committee. Details of treatment protocols POG-9421, CCG-2891, and CCG-2961 have been described previously.16,–18 COG AAML03P1 was a pilot study in which patients with de novo AML received gemtuzumab ozogamicin, a humanized immunoglobulin G4 anti-CD33 monoclonal antibody linked to the cytotoxic agent calicheamicin, in combination with a backbone of Medical Research Council 12–based conventional chemotherapy.19 Cytogenetic data were available for 84%, 53%, 62%, and 93% of patients enrolled on POG-9421, CCG-2891, CCG-2961, and COG AAML03P1, respectively. A diagnosis of CBF AML was made if either t(8;21) (q22;22) or inv (16) (p13;q22)/t(16,16)(p13;q22) was detected in pretreatment bone marrow (BM) or peripheral blood (PB) specimens by either conventional cytogenetics or fluorescent in situ hybridization or both techniques. In most cases chromosomal abnormalities were identified by conventional cytogenetics, and karyotypes were confirmed by central review (S.C.R. for POG 9421; N.A.H. for CCG-2891 and CCG-2961; S.C.R. and B.H. for COG AAML03P1).
KIT mutational analysis
Genomic DNA was extracted from diagnostic BM or PB specimens with the use of the Puregene Protocol (Gentra Systems). KIT exons 8 and 17 were amplified by genomic polymerase chain reaction (PCR) in separate reactions.
PCR amplification of KIT exon 17
PCR amplification of exon 17 was performed with primers 17F, 5′-CCTCCAACCTAATAGTGTATTCACAG, and 17R, 5-ATGTGTGATATCCCTAGACAGGAT. The PCR mixture contained a maximum of 100 ng of DNA and 10 pmol/μL 17F and17R primers. Standard concentrations of reaction mixtures were used as described previously.20 Negative controls were included with amplification. Denaturing, annealing, and extension steps were performed at 95°C for 30 seconds, 61°C for 25 seconds, and 72°C for 25 seconds, respectively, for a total of 40 cycles on an MJ Research thermocycler. An initial 5-minute denaturation step at 95°C and a final 8-minute extension step at 72°C were also performed. PCR products were resolved on a 2% agarose gel. After visual confirmation of amplification, 4 μL of the PCR product was purified with 2 μL of ExoSAP-IT (US Biochemical Corp) and analyzed by bidirectional sequencing on an ABI 377 sequencer, using the BigDye terminator kit (Applied Biosystems Inc). Mutational analysis was conducted by Mutation Surveyor (SoftGenetics).
PCR amplification of KIT exon 8
PCR amplification of exon 8 was performed with primers 8F, 5′-TTCAGATTCTGCCCTTTGAACTTG, and 8R, 5-TGAAATTCAAGTGAATTGCAGTCC, with reaction conditions identical to those for exon 17 analysis. The 8R primer was labeled with a 5′ fluorescent probe to facilitate mutation detection by microcapillary electrophoresis techniques. PCR products were resolved on a 2% agarose gel and analyzed by the Genescan or Genemapper software (Applied Biosystems Inc) after appropriate dilution of PCR products to facilitate identification of mutational insertions or deletions or both. Mutations were confirmed with the use of the aforementioned sequencing techniques when possible.
Denaturing high-performance liquid chromatography analysis of samples from patients enrolled on CCG-2961
A subset of patient samples obtained from CCG-2961 were also subjected to exon 8 and exon 17 mutational analysis by the Transgenomic WAVE denaturing high-performance liquid chromatography system (Transgenomic Inc) with the use of previously described methods.12
Statistical methods
Clinical outcome data from POG-9421, CCG-2891, CCG-2961, and COG AAML03P1 were analyzed through September 5, 2006, January 14, 2004, October 30, 2006, and August 28, 2008, respectively. The median follow-up for all eligible patients with de novo AML alive at last contact for the clinical trials included in our analysis is 2003.5 days (range, 56-3136 days) for POG-9421, 3128 days (range, 1-5028 days) for CCG-2891, and 1762.5 days (range, 0-3620 days) for CCG-2961. Median days of follow-up for COG AAML03P1 is 783 days (range, 0-1456 days). For the purpose of this study, patients were defined as being in complete remission (CR) if they had 5% or fewer blasts and trilineage recovery after 2 courses of chemotherapy. OS was defined as the time from study entry until death. Event-free survival (EFS) was defined as the time from study entry until death, induction failure, or relapse. Disease-free survival (DFS) was defined as the time from induction CR until relapse or death, and relapse risk (RR) was defined as the time from the end of induction to relapse or death as a result of progressive disease, whereby deaths from nonprogressive disease were considered competing events. Pearson chi-square test was used to test for differences in the distribution of categorical variables. The Fisher exact test was used when data were sparse. The Mann-Whitney test was used to analyze differences of medians.21 The Kaplan-Meier method was used for nonparametric survival curve analyses for OS, EFS, and DFS.22 RR analyses were performed with the use of methods of cumulative incidence. Patients having a matched family donor were censored at the end of 2 courses of therapy for OS, EFS, DFS, and RR. Patients lost to follow-up were censored at their date of last known contact or at a cutoff of 6 months before the frozen date of study data to compensate for the tendency of deaths and relapses to be reported sooner than ongoing follow-up. Differences in OS, EFS, and DFS were tested with the use of Cox proportional hazards models,23 which were stratified by the protocol on which participants were treated to account for any differences in treatment protocols. Differences between cumulative incidence curves were tested with the Gray test24 stratified by the study on which participants were enrolled. Confidence intervals for survival estimates were calculated with Greenwood estimate of the standard error.25 Binomial confidence intervals (CIs) were also provided for prevalence proportions.
Results
Study population
All patients with de novo CBF AML enrolled on POG-9421, CCG-2891, CCG-2961, and COG-AAML03P1 were eligible for this study. Forty-eight (49%) of 97, 18 (20%) of 89, 98 (71%) of 138, and 39 (50%) of 78 eligible patients with CBF AML from POG-9421, CCG-2891, CCG-2961, and COG-AAML03P1, respectively, had diagnostic BM or PB specimens available for KIT analysis. In total, 203 (50%) of 402 CBF AML patient samples were analyzed.
Patients and treatment
To determine whether our study population was representative of the overall CBF AML population, we compared laboratory and clinical characteristics of the 203 study patients with those of the remaining 199 patients with CBF AML who lacked samples for analysis. Patients included in our study had a significantly higher median white blood cell count (on study 28 800 × 109/L on study vs 19 600 × 109/L off study; P = .036) and median age (11.8 years on study vs 9.4 years off study; P = .008) than patients for whom samples were unavailable. Percentage of BM blasts at presentation was higher but not significantly different for our study population (52.5%) than for patients off study (46.5%; P = .117). Moreover, our study population had fewer males (52%) than the population excluded from analysis (61%); this finding approached statistical significance (P = .066). There were no significant differences in 5-year OS (74% ± 7% on study vs 71% ± 8% off study; hazard ratio [HR] = 1.1; P = .685) and EFS (58% ± 8% on study vs 55% ± 9% off study; HR = 1.1; P = .628) for the 2 groups. DFS (59% ± 9% on study vs 59% ± 10% off study; HR = 0.98; P = .906) and RR (35% ± 8% on study vs 38% ± 10% off study; HR = 1.02; P = .830) were also comparable. Clinical outcome of all patients with CBF AML (regardless of KIT mutational status or inclusion in our study) was also compared with that of patients without CBF AML with known cytogenetic information. Consistent with previous studies, we found that 5-year OS, EFS, DFS, and RR was superior for those patients with CBF AML (5-year OS: 73% ± 5% for CBF vs 43% ± 3% for non-CBF, HR = 0.38, P < .001; 5-year EFS: 57% ± 6% for CBF vs 33% ± 3% for non-CBF, HR = 0.49, P < .001; 5-year DFS: 59% ± 6% for CBF vs 40% ± 3% for non-CBF, HR = 0.58, P < .001; 5-year RR: 36% ± 6% for CBF vs 55% ± 4% for non-CBF, HR = 0.55, P < .001).
Prevalence and type of KIT mutations at diagnosis
KIT mutations were detected in 38 (19%; 95% CI, 14%-25%) of the 203 specimens for which KIT mutational analysis (exon 8 and exon 17) was performed. Prevalence of KIT mutations ranged from 15% to 28% on individual cooperative studies. Of the 38 mutations detected, 20 (52.5%; 95% CI, 36%-69%) involved exon 8, 17 (45%; 95% CI, 29%-62%) involved exon 17, and 1 (2.5%; 95% CI, 0%-14%) affected both regions (Table 1). Exon 8 mutations were either small deletions or insertions detected in 5% (95% CI, 2%-11%) of t(8;21) and 16% (95% CI, 9%-25%) of inv(16) patient samples (Table 1). All sequenced samples (13 of 20) had deletions or insertions involving codons 416-420. Exon 17 mutations were exclusively point mutations and were found in 12% (95% CI, 6%-19%) of t(8;21) and 4% (95% CI, 1%-11%) of inv(16) patient samples (Table 1). Twelve of 17 exon 17 mutations involved codon 816 (n = 9 D816V, n = 1 D816H, n = 2 D816Y) and 5 involved surrounding codons (n = 1 D820G, n = 4 N822K). Mutations of both exon 8 (insertion at codon 418) and exon 17 (D816Y) were detected in 1 inv(16) patient sample. Of the 83 samples screened by denaturing high-performance liquid chromatography, all previously identified KIT mutations were confirmed, but no additional mutations were detected.
Prevalence and type of KIT mutations in diagnostic specimens from 203 patients with CBF AML enrolled on 4 pediatric oncology trials
| No. of samples analyzed | ||||||
|---|---|---|---|---|---|---|
| Total | WT KIT, n (%) | KIT mutation+, n (%) | Exon 8 mutation+, no. (%) | Exon 17 mutation+, no. (%) | Both exon 8/17 mutation+, no. (%) | |
| Summation of all studies | ||||||
| All CBF AML | 203 | 165 (81) | 38 (19) | 20 (10) | 17 (8) | 1 (< 1) |
| t(8;21) | 113 | 94 (83) | 19 (17) | 6 (5) | 13 (12) | 0 (0) |
| inv(16) | 90 | 71 (79) | 19 (21) | 14 (16) | 4 (4) | 1 (1) |
| By study | ||||||
| POG-9421 | ||||||
| All CBF AML | 48 | 37 (77) | 11 (23) | 5 (10) | 6 (13) | 0 (0) |
| t(8;21) | 25 | 17 (68) | 8 (32) | 3 (12) | 5 (20) | 0 (0) |
| inv(16) | 23 | 20 (87) | 3 (13) | 2 (9) | 1 (4) | 0 (0) |
| CCG-2891 | ||||||
| All CBF AML | 18 | 13 (72) | 5 (28) | 4 (22) | 1 (6) | 0 (0) |
| t(8;21) | 8 | 7 (88) | 1 (13) | 0 (0) | 1 (13) | 0 (0) |
| inv(16) | 10 | 6 (60) | 4 (40) | 4 (40) | 0 (0) | 0 (0) |
| CCG-2961 | ||||||
| All CBF AML | 98 | 83 (85) | 15 (15) | 8 (8) | 6 (6) | 1 (1) |
| t(8;21) | 61 | 53 (87) | 8 (13) | 3 (5) | 5 (8) | 0 (0) |
| inv(16) | 37 | 30 (81) | 7 (19) | 5 (14) | 1 (3) | 1 (3) |
| COG AAML03P1 | ||||||
| All CBF AML | 39 | 32 (82) | 7 (18) | 3 (8) | 4 (10) | 0 (0) |
| t(8;21) | 19 | 17 (89) | 2 (11) | 0 (0) | 2 (11) | 0 (0) |
| inv(16) | 20 | 15 (75) | 5 (25) | 3 (15) | 2 (10) | 0 (0) |
| No. of samples analyzed | ||||||
|---|---|---|---|---|---|---|
| Total | WT KIT, n (%) | KIT mutation+, n (%) | Exon 8 mutation+, no. (%) | Exon 17 mutation+, no. (%) | Both exon 8/17 mutation+, no. (%) | |
| Summation of all studies | ||||||
| All CBF AML | 203 | 165 (81) | 38 (19) | 20 (10) | 17 (8) | 1 (< 1) |
| t(8;21) | 113 | 94 (83) | 19 (17) | 6 (5) | 13 (12) | 0 (0) |
| inv(16) | 90 | 71 (79) | 19 (21) | 14 (16) | 4 (4) | 1 (1) |
| By study | ||||||
| POG-9421 | ||||||
| All CBF AML | 48 | 37 (77) | 11 (23) | 5 (10) | 6 (13) | 0 (0) |
| t(8;21) | 25 | 17 (68) | 8 (32) | 3 (12) | 5 (20) | 0 (0) |
| inv(16) | 23 | 20 (87) | 3 (13) | 2 (9) | 1 (4) | 0 (0) |
| CCG-2891 | ||||||
| All CBF AML | 18 | 13 (72) | 5 (28) | 4 (22) | 1 (6) | 0 (0) |
| t(8;21) | 8 | 7 (88) | 1 (13) | 0 (0) | 1 (13) | 0 (0) |
| inv(16) | 10 | 6 (60) | 4 (40) | 4 (40) | 0 (0) | 0 (0) |
| CCG-2961 | ||||||
| All CBF AML | 98 | 83 (85) | 15 (15) | 8 (8) | 6 (6) | 1 (1) |
| t(8;21) | 61 | 53 (87) | 8 (13) | 3 (5) | 5 (8) | 0 (0) |
| inv(16) | 37 | 30 (81) | 7 (19) | 5 (14) | 1 (3) | 1 (3) |
| COG AAML03P1 | ||||||
| All CBF AML | 39 | 32 (82) | 7 (18) | 3 (8) | 4 (10) | 0 (0) |
| t(8;21) | 19 | 17 (89) | 2 (11) | 0 (0) | 2 (11) | 0 (0) |
| inv(16) | 20 | 15 (75) | 5 (25) | 3 (15) | 2 (10) | 0 (0) |
CBF AML indicates core binding factor acute myeloid leukemia; WT, wild-type; +, mutation positive; POG, Pediatric Oncology Group; CCG, Children's Cancer Group; and COG, Children's Oncology Group.
Clinical characteristics and CR rates for patients with KIT mutations
Clinical characteristics and CR rates for the 38 patients with KIT mutations were compared with those of the 165 patients with CBF AML with wild-type (WT) KIT. There were no significant differences in sex, race, and median age between the 2 groups although more patients with KIT mutations were younger than 2 years of age (P = .077; Table 2). Median diagnostic white blood cell count, median BM blast percentage at diagnosis, and CR rates were also similar for both groups (Table 2).
Clinical characteristics and CR rates for patients with CBF AML with and without KIT mutations
| KIT mutation+ | WT KIT | P | |
|---|---|---|---|
| Total, n | 38 | 165 | |
| Sex | |||
| Male, n (%) | 22 (58) | 84 (51) | .437 |
| Female, n (%) | 16 (42) | 81 (49) | |
| Age, y | |||
| Median (range) | 10.5 (1.1-16.6) | 12.0 (0.6-19.6) | .127 |
| 0 to younger than 2 y, n (%) | 4 (11) | 6 (4) | .077 |
| 2 to younger than 10 y, n (%) | 14 (37) | 55 (33) | .681 |
| 10-21 y, n (%) | 20 (53) | 104 (63) | .236 |
| Race | |||
| White, n (%) | 23 (61) | 108 (66) | .504 |
| Nonwhite, n (%) | 15 (39) | 55 (34) | |
| Unknown, n | 0 | 2 | |
| WBC count, median (range) | 33.5 (3.9-379) | 27.1 (1.6-373) | .477 |
| BM blasts, %, median (range) | 56 (0-92) | 52 (0-99) | .915 |
| Additional mutations | |||
| FLT3/ITD | |||
| Negative, n (%) | 31 (100) | 139 (96) | .592 |
| Positive, n (%) | 0 (0) | 6 (4) | |
| NPM1 | |||
| Negative, n (%) | 17 (100) | 110 (100) | > .999 |
| Positive, n (%) | 0 (0) | 0 (0) | |
| CEBPA | |||
| Negative, n (%) | 21 (100) | 103 (100) | > .999 |
| Positive, n (%) | 0 (0) | 0 (0%) | |
| WT-1 | |||
| Negative, n (%) | 22 (100) | 96 (88) | .124 |
| Positive, n (%) | 0 (0) | 13 (12) | |
| BM blasts, %, median (range) | 56 (0-92) | 52 (0-99) | .195 |
| Induction response | |||
| CR, n (%) | 36 (97) | 144 (94) | .690 |
| Not in CR, n (%) | 1 (3) | 9 (6) | |
| Unevaluable or withdrew, n | 1 | 12 |
| KIT mutation+ | WT KIT | P | |
|---|---|---|---|
| Total, n | 38 | 165 | |
| Sex | |||
| Male, n (%) | 22 (58) | 84 (51) | .437 |
| Female, n (%) | 16 (42) | 81 (49) | |
| Age, y | |||
| Median (range) | 10.5 (1.1-16.6) | 12.0 (0.6-19.6) | .127 |
| 0 to younger than 2 y, n (%) | 4 (11) | 6 (4) | .077 |
| 2 to younger than 10 y, n (%) | 14 (37) | 55 (33) | .681 |
| 10-21 y, n (%) | 20 (53) | 104 (63) | .236 |
| Race | |||
| White, n (%) | 23 (61) | 108 (66) | .504 |
| Nonwhite, n (%) | 15 (39) | 55 (34) | |
| Unknown, n | 0 | 2 | |
| WBC count, median (range) | 33.5 (3.9-379) | 27.1 (1.6-373) | .477 |
| BM blasts, %, median (range) | 56 (0-92) | 52 (0-99) | .915 |
| Additional mutations | |||
| FLT3/ITD | |||
| Negative, n (%) | 31 (100) | 139 (96) | .592 |
| Positive, n (%) | 0 (0) | 6 (4) | |
| NPM1 | |||
| Negative, n (%) | 17 (100) | 110 (100) | > .999 |
| Positive, n (%) | 0 (0) | 0 (0) | |
| CEBPA | |||
| Negative, n (%) | 21 (100) | 103 (100) | > .999 |
| Positive, n (%) | 0 (0) | 0 (0%) | |
| WT-1 | |||
| Negative, n (%) | 22 (100) | 96 (88) | .124 |
| Positive, n (%) | 0 (0) | 13 (12) | |
| BM blasts, %, median (range) | 56 (0-92) | 52 (0-99) | .195 |
| Induction response | |||
| CR, n (%) | 36 (97) | 144 (94) | .690 |
| Not in CR, n (%) | 1 (3) | 9 (6) | |
| Unevaluable or withdrew, n | 1 | 12 |
CBF AML indicates core binding factor acute myeloid leukemia; WT, wild-type; WBC, white blood cell; BM, bone marrow; FLT3/ITD, FLT3 internal tandem duplication; NPM1, nucleophosmin; CEBPA, CCAAT/enhancer binding protein-α; WT-1, Wilms tumor gene 1; and CR, complete remission.
A significant number of our CBF study samples had been previously screened for other AML-related molecular abnormalities.26,,,–30 No additional mutations were detected in the KIT mutation–positive cohort. Specifically, although FLT3 internal tandem duplications (FLT3/ITD) and WT-1 mutations were observed in 6 and 13 patients with CBF AML, respectively, these mutations were limited to patients with WT KIT CBF AML. CEBPA and nucleophosmin (NPM1) mutations were absent in all CBF samples analyzed (Table 2).
Prognostic significance of KIT mutations in CBF AML
Five-year OS and EFS from study entry (note that 3-year OS/EFS rates were used for COG protocol AAML03P1 because of the lack of follow-up) was similar for patients with CBF AML with and without KIT mutations (Table 3; Figure 1). The 5-year cumulative risk of relapse and corresponding DFS from CR were also similar for the 2 groups (Table 3; Figure 2). There were no significant differences in outcome for patients with KIT mutations or WT KIT within individual studies with the exception of POG-9421, wherein 11 patients with KIT mutations had poorer 5-year OS but similar 5-year EFS than patients with WT KIT (Table 3).
Outcome data for patients with CBF AML with and without KIT mutations
| KIT mutation | WT KIT | P | |||
|---|---|---|---|---|---|
| (N = 38) | Outcome data | (N = 165) | Outcome data | ||
| N | % ± 2SE % | N | % ± 2SE % | ||
| 5-y OS from study entry | |||||
| All studies | 38 | 76 ± 15 | 165 | 74 ± 8 | .819 |
| POG-9421 | 11 | 64 ± 29 | 37 | 89 ± 12 | .014 |
| CCG-2891 | 5 | 100 ± 0 | 13 | 67 ± 38 | .577 |
| CCG-2961 | 15 | 79 ± 22 | 83 | 68 ± 12 | .701 |
| COG AAML03P1* | 7 | 67 ± 54 | 32 | 77 ± 20 | .589 |
| 5-y EFS from study entry | |||||
| All studies | 38 | 55 ± 17 | 165 | 59 ± 9 | .857 |
| POG-9421 | 11 | 55 ± 30 | 37 | 66 ± 18 | .563 |
| CCG-2891 | 5 | 80 ± 36 | 13 | 53 ± 41 | .925 |
| CCG-2961 | 15 | 57 ± 26 | 83 | 57 ± 12 | .866 |
| COG AAML03P1* | 7 | 44 ± 44 | 32 | 61 ± 21 | .518 |
| 5-y RR from end of induction | |||||
| All studies | 36 | 35 ± 17 | 144 | 34 ± 10 | .877 |
| POG-9421 | 11 | 36 ± 29 | 36 | 35 ± 19 | .961 |
| CCG-2891 | 4 | 0 ± 0 | 13 | 50 ± 41 | .255 |
| CCG-2961 | 14 | 31 ± 26 | 66 | 33 ± 14 | .814 |
| COG AAML03P1* | 7 | 56 ± 44 | 29 | 30 ± 21 | .512 |
| 5-y DFS from induction CR | |||||
| All studies | 36 | 56 ± 18 | 144 | 60 ± 10 | .786 |
| POG-9421 | 11 | 55 ± 30 | 36 | 65 ± 19 | .58 |
| CCG-2891 | 4 | 100 ± 0 | 13 | 50 ± 41 | .541 |
| CCG-2961 | 14 | 54 ± 28 | 66 | 58 ± 14 | .707 |
| COG AAML03P1* | 7 | 44 ± 44 | 29 | 61 ± 22 | .484 |
| KIT mutation | WT KIT | P | |||
|---|---|---|---|---|---|
| (N = 38) | Outcome data | (N = 165) | Outcome data | ||
| N | % ± 2SE % | N | % ± 2SE % | ||
| 5-y OS from study entry | |||||
| All studies | 38 | 76 ± 15 | 165 | 74 ± 8 | .819 |
| POG-9421 | 11 | 64 ± 29 | 37 | 89 ± 12 | .014 |
| CCG-2891 | 5 | 100 ± 0 | 13 | 67 ± 38 | .577 |
| CCG-2961 | 15 | 79 ± 22 | 83 | 68 ± 12 | .701 |
| COG AAML03P1* | 7 | 67 ± 54 | 32 | 77 ± 20 | .589 |
| 5-y EFS from study entry | |||||
| All studies | 38 | 55 ± 17 | 165 | 59 ± 9 | .857 |
| POG-9421 | 11 | 55 ± 30 | 37 | 66 ± 18 | .563 |
| CCG-2891 | 5 | 80 ± 36 | 13 | 53 ± 41 | .925 |
| CCG-2961 | 15 | 57 ± 26 | 83 | 57 ± 12 | .866 |
| COG AAML03P1* | 7 | 44 ± 44 | 32 | 61 ± 21 | .518 |
| 5-y RR from end of induction | |||||
| All studies | 36 | 35 ± 17 | 144 | 34 ± 10 | .877 |
| POG-9421 | 11 | 36 ± 29 | 36 | 35 ± 19 | .961 |
| CCG-2891 | 4 | 0 ± 0 | 13 | 50 ± 41 | .255 |
| CCG-2961 | 14 | 31 ± 26 | 66 | 33 ± 14 | .814 |
| COG AAML03P1* | 7 | 56 ± 44 | 29 | 30 ± 21 | .512 |
| 5-y DFS from induction CR | |||||
| All studies | 36 | 56 ± 18 | 144 | 60 ± 10 | .786 |
| POG-9421 | 11 | 55 ± 30 | 36 | 65 ± 19 | .58 |
| CCG-2891 | 4 | 100 ± 0 | 13 | 50 ± 41 | .541 |
| CCG-2961 | 14 | 54 ± 28 | 66 | 58 ± 14 | .707 |
| COG AAML03P1* | 7 | 44 ± 44 | 29 | 61 ± 22 | .484 |
CBF AML indicates core binding factor acute myeloid leukemia; WT, wild-type; OS, overall survival; POG, Pediatric Oncology Group; CCG, Children's Cancer Group; COG, Children's Oncology Group; EFS, event-free survival; RR, relapse risk; and DFS, disease-free survival.
Three-year estimates are used because of lack of follow-up.


We also evaluated the prognostic significance of KIT mutation type (exon 8 vs exon 17) for this CBF AML cohort. Rates of induction CR for patients with exon 8 or exon 17 mutations or both mutations were comparable to those of patients with WT KIT (Table 4). Five-year EFS and OS from study entry were also similar regardless of KIT mutation status as was corresponding 5-year DFS and RR from CR (Table 4).
Clinical outcomes for patients with CBF AML with KIT mutations (exon 8, exon 17, or both) compared with patients with WT KIT
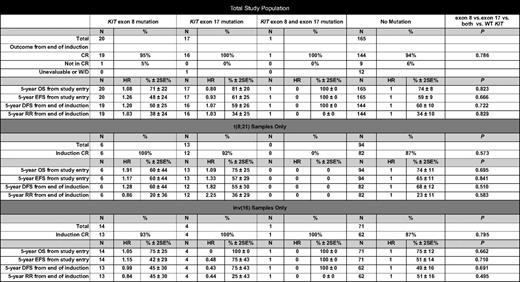
Data are presented for the entire study population and by cytogenetic subtype. CBF AML indicates core binding factor acute myeloid leukemia; WT, wild-type; CR, complete remission; W/D, withdrawn; HR, hazard ratio; SE, standard error; OS, overall survival; EFS, event-free survival; DFS, disease-free survival; and RR, relapse risk.
Patients with t(8;21) AML and KIT mutations of exon 17 codon 816 have been found to have a particularly unfavorable prognosis in previous studies.7,9,10 In our cohort, 12 patients [8 t(8;21) and 4 inv(16) AML] had mutations in codon 816 (n = 9 D816V, n = 1 D816H, n = 2 D816Y). Although small sample size precluded subanalysis by cytogenetic type, patients with CBF AML and KIT D816 mutations (KIT D816) had similar CR rates compared with those patients with other KIT mutations or WT KIT (KIT D816, 100%; other KIT mutations, 96%; WT KIT, 94%; P = .649). OS from study entry (KIT D816, 82% ± 23%; other KIT mutations, 74% ± 18%; WT KIT, 74% ± 8%; P = .965) and EFS from study entry (KIT D816, 61% ± 33%; other KIT mutations, 52% ± 21%; WT KIT, 59% ± 9%; P = .894) were also comparable for the 3 groups. Five-year DFS (KIT D816, 62% ± 30%; other KIT mutations, 52% ± 21%; WT KIT, 60% ± 10%; P = .890) and RR from end of induction (KIT D816, 29% ± 28%; other KIT mutations, 38% ± 21%; WT KIT, 34% ± 10%; P = .882) were also similar.
Prognostic significance of KIT mutations for patients with t(8;21).
Of the 113 patients with t(8;21) AML, 19 (17%; 95% CI, 10%-25%) had a KIT mutation (6 involved exon 8 and 13 involved exon 17; Table 1). CR rates for patients with t(8;21) AML were similar regardless of KIT mutation status (100% for KIT mutation vs 92% for WT KIT; P = .599). The 5-year OS (71% ± 22% for KIT mutation vs 74% ± 11% for WT KIT; P = .603) and EFS from study entry (58% ± 24% for KIT mutation vs 65% ± 11% for WT KIT; P = .572) as well as 5-year DFS (64% ± 25% for KIT mutation vs 75% ± 12% for WT KIT; P = .293) and RR from induction CR (31% ± 23% for KIT mutation vs 23% ± 11% for WT KIT; P = .516) were similar for both groups. Location of the mutation did not affect outcome. CR rates for patients with t(8;21) AML with KIT exon 8 or exon 17 mutations were comparable to those with WT KIT (Table 4). Five-year EFS and OS rates from study entry as well as 5-year DFS and RR rates from end of induction were also similar regardless of KIT genotype (Table 4).
Prognostic significance of KIT mutations for patients with inv(16).
Of the 90 patients with inv(16) AML, 19 (21%; 95% CI, 13%-31%) had KIT mutations (14 involved exon 8, 4 involved exon 17, and 1 involved both exons; Table 1). CR rates for patients with inv(16) AML with or without KIT mutations were similar (95% for KIT mutation vs 97% for WT KIT; P = .547). Rates of 5-year OS (81% ± 19% for KIT mutation vs 75% ± 12% for WT KIT; P = .607) and EFS from study entry (53% ± 25% for KIT mutation vs 51% ± 14% for WT KIT; P = .770) were also comparable for both groups. DFS and RR rates from induction CR did not differ for both groups, although outcomes were slightly better for patients with KIT mutations than for patients with WT KIT. (DFS: 56% ± 25% for KIT mutation vs 49% ± 16% for WT KIT, P = .528; RR: 38% ± 25% for KIT mutation vs 51% ± 16% for WT KIT, P = .295). Location of mutation did not affect outcome. CR rates for patients with inv(16) AML with KIT exon 8 or 17 mutations, or both, were similar to those without mutations (Table 4). Five-year EFS and OS rates from study entry as well as 5-year DFS and RR rates from end of induction were also similar regardless of KIT genotype (Table 4).
Discussion
We retrospectively examined the prevalence and prognostic significance of KIT exon 8 and 17 mutations in a cohort of 203 children with CBF AML. This study of patients with CBF AML enrolled on 4 pediatric cooperative AML trials is the largest KIT mutational analysis of this subpopulation to date. The prevalence of KIT mutations was approximately 20% in our study population, which is in the range reported by previous studies of adults (6%-48%)2,6,,,,–11 and children (17%-41%)12,,–15 with CBF AML. We found that KIT mutations lacked prognostic significance in pediatric patients with CBF AML. This finding is in contrast to that of previous studies of adults with CBF AML in which KIT mutations predicted a higher rate of relapse,2,6,10 OS,2,9,–11 EFS,9,11 and DFS11 in particular analyses. Specifically, Care et al6 showed that mutations of exon 8 in adults with inv(16) AML adversely affected relapse rate but not OS, whereas Cairoli et al10 and Boissel et al11 found that KIT mutations lacked prognostic significance in adult patients with inv(16) CBF AML. A later series by Paschka et al2 found that adult patients with inv(16) CBF AML had a higher risk of relapse. Strikingly, RR rates for patients with inv(16) with mutations in exon 17 were more than 6 times higher than for patients without KIT mutations. Mutations in exon 17, exon 8, or both also negatively affected OS.2
The prognostic effect of KIT mutations in t(8;21) AML is also debatable. Both Schnittger et al9 and Cairoli et al10 found that exon 17 mutations negatively affected outcome. In the former study, D816 mutations were exclusively evaluated and adversely affected EFS and OS.9 In the latter study, patients samples were screened for mutations in exon 17, 8, and 11 and found, by subset analysis, that only exon 17 mutations had prognostic significance.10 Boissel et al11 also showed that KIT mutations in t(8;21) AML negatively affected OS, EFS, and relapse-free survival, although analysis of outcome by specific genotype was not performed. Paschka et al2 found that patients with KIT mutations in t(8;21) had 5 times higher risk of relapse, but similar rates of OS to, than patients without KIT mutations. Nine of 11 mutations detected involved exon 17.
The prognostic significance of KIT mutations in pediatric CBF AML has varied in previously published series. Goemans et al12 identified KIT exon 8 and 17 mutations in 10 (37%) of 27 children with CBF AML, with an overall prevalence of 55% for inv(16) (27% for exon 8, 27% for exon 17) and 31% (12.5% for exon 8, 18.8% for exon 17) for children with t(8;21) AML. In their study, the presence of a KIT mutations was not associated with inferior EFS.12 Shih et al15 detected KIT mutations in 17 (41%) of 41 patients with CBF AML [5 (29%) involved exon 8, 9 (53%) involved exon 17, and 2 involved mutations at both exon 8 and 17]. No significant difference in OS or EFS was observed for patients with KIT mutations compared with WT KIT. Analysis by cytogenetic subtype also showed that patients with KIT mutations and patients with WT KIT had similar EFS compared with patients with WT KIT.15 The finding of Goemans et al12 and Shih et al15 are in contrast to a study by Shimada et al.13 In this study, 46 children with t(8;21) AML were screened for KIT mutations of exon 8, 9, 10, 11, 17, and 18. Eight (17.4%) of 46 patients had KIT mutations, all involving the second intracellular kinase domain (exon 17 and 18). Those patients with KIT mutations had lower OS and DFS and increased RR than patients without KIT abnormalities.13
The prevalence of KIT exon 8 or 17 mutations in our study cohort (19%) is lower than that reported by Goemans et al12 and Shih et al15 but comparable to that of an analysis by Shimada et al13,14 that was limited to patients with t(8;21) AML. Exon 8 mutations were observed more frequently in patients with inv(16) AML [74% versus 32% of t(8;21) samples analyzed], whereas mutations of exon 17 were more prevalent in t(8;21) AML [68% versus 21% of inv(16) samples analyzed], consistent with published findings.5,9 Like previous studies, we also found that exon 17 mutations associated with inv(16) disease occurred exclusively at codon D816,2,6,9,10 whereas mutations associated with t(8;21) occurred predominantly at codons D816 and N822.2,7,8
Our study of 4 large cooperative pediatric trials highlights that KIT mutations lack prognostic significance within pediatric CBF AML. Our findings, which differ from several adult series, may reflect inherent biologic differences between KIT mutations in children and adults with CBF AML.2,6,9,–11 In vitro studies have shown that exon 8 mutations represent a gain of function mutation that induces KIT receptor hyperactivation in response to stem cell factor stimulation. This results in excessive proliferation and resistance to apoptotic cell death.31 In contrast, exon 17 mutations promote receptor autophosphorylation without the requirement of receptor–ligand interaction. Such mutations result in constitutive activation of phosphatidylinositol 3 kinase and its downstream targets as well as STAT3.5,32,33 Although the types of exon 8 and 17 mutations observed in adult and pediatric CBF AML are similar, it is possible that the prognostic implications observed in adults may reflect a tendency for the mutation to achieve clonal dominance in a less mature leukemic progenitor that is more difficult to target with conventional chemotherapies. We have previously demonstrated an association between lineage involvement of FLT3/ITD mutations in pediatric AML and clinical response to therapy. In that study, pediatric patients with FLT3/ITD disease limited to CD34+/CD33+ progenitors had markedly improved outcome compared with patients with CD34+/CD33− FLT3/ITD involvement.34 A similar paradigm may exist for pediatric versus adult patients with CBF AML with KIT mutations. Alternatively, KIT mutations in adults may evolve from a backbone of greater genetic instability and be associated with additional unidentified mutations that confer prognostic significance/greater resistance to conventional agents. Additional studies aimed at defining the maturational stage of hematopoietic development at which these mutations achieve dominance in adult versus pediatric patients with CBF AML may be illustrative. Alternatively, with the increased use of whole genome arrays, identification of associated mutations that confer poor outcome in KIT mutation–positive CBF AML may be detected.
The lack of clinical prognostic significance observed for KIT mutations in pediatric compared with adult CBF AML may also reflect the intense nature of pediatric therapeutic regimens. The outcomes of our study cohort are similar to those of 2 smaller pediatric series12,15 but contrast with that observed by Shimada et al.13 In that study, KIT WT t(8;21) AML outcomes were quite high (EFS, 92%)13 compared with that of similar patients in both our series (WT KIT EFS, 59%) and that of the Europeans (WT KIT EFS, 63%12,13 ), which may explain the inferior outcome observed comparing patients with KIT mutation positive with patients with WT KIT within that cohort. We concede that patients included in our analyses were treated in 4 different clinical trials, which may have introduced an error in analysis of the combined data. However, although clinical outcomes varied from study to study, the prognostic implications of KIT mutations remained consistent in each trial.
In childhood AML, most patients with a matched family donor (MFD) undergo allogeneic hematopoietic stem cell transplantation in first CR. Children with CBF AML in first CR have been considered a more favorable risk group than non-CBF AML, given their superior OS and DFS.35 Moreover, their OS and DFS has not improved with MFD hematopoietic stem cell transplantation.35 These patients are, therefore, treated solely with chemotherapy even if an MFD is available. Because our study shows that pediatric patients with CBF AML with KIT mutations have similar outcome to patients with CBF AML without such mutations, their presence does not provide rationale for alteration of therapy. This finding is in contrast to adult CBF AML, whereby the inferior outcome observed with KIT mutations may justify therapy modification. The utility of receptor tyrosine kinase inhibitors for this patient population remains uncertain. Anecdotal use of imatinib, both as a single agent or in combination with conventional chemotherapy, has been partly effective in patients with exon 8 but not exon 17 D816 mutations, consistent with results from in vitro analysis.2,8,36,–38 The use of second-generation tyrosine kinase inhibitors, such as dasatinib or nilotinib, may broaden the number of mutation types that can be targeted.39,40 Ultimately, their role in treatment of pediatric patients with CBF AML with KIT mutations warrants further evaluation in cooperative pediatric clinical trials.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Ms Ling Pan for her technical contribution to this work, Vani J. Shanker, PhD, for her scientific editing, and the COG AML Reference Laboratory, particularly Sommer Castro, for providing diagnostic specimens. We thank the patients and families who consented to the use of biologic specimens in these trials.
This work was supported by the National Cancer Institute (5K12CA076930-08) (J.A.P.), St Baldrick's Foundation Career Development Award (J.A.P.), CureSearch Research Fellowship Award (J.A.P.), For Julie Foundation Research Grant (J.A.P.), VA Merit Review Grant (M.C.H.), Leukemia & Lymphoma Society (M.C.H.), and the National Institutes of Health (grants R21 CA10262-01 and R01 CA114563-01, Children's Oncology Group Chair's grant NIH U10 CA98543).
National Institutes of Health
Authorship
Contribution: J.A.P. designed and performed research, analyzed data, and wrote the manuscript; T.A.A. (senior statistician) performed statistical analysis and edited the manuscript; R.B.G. performed statistical analysis and edited the manuscript; R.Z. performed research, analyzed data, and edited the manuscript; P.A.H., Y.R., G.D., N.J.L., D.B., M.C., H.J.W., B.H., S.C.R., N.A.H., R.J.A., J.P.R., and I.D.B. analyzed data and edited the manuscript; W.G.W., B.J.L., and C.H. (clinical study principal investigators) edited the manuscript; and M.C.H. and S.M. designed research, analyzed data, and edited the manuscript.
Conflict-of-interest disclosure: M.C.H. has an equity interest in MolecularMD, a molecular diagnostic company that performs molecular testing of leukemia samples, and serves as an unpaid consultant for the company. His conflict of interest is managed by Oregon Health and Science University and Portland Veterans Affairs Medical Center Conflict of Interest in Research Committees. The remaining authors declare no competing financial interests.
Correspondence: Jessica Pollard, Seattle Children's Hospital, 4800 Sandpoint Way NE, Division of Heme/Onc, MS-B-6553, Seattle, WA 98105; e-mail: jessica.pollard@seattlechildrens.org.
