

The clonal CD3−CD4+ T-cell population characterizing lymphocytic variant hypereosinophilic syndrome (L-HES) persists for years, with a subgroup of patients ultimately progressing to T lymphoma. The molecular changes associated with the premalignant clone and the emergence of malignant subclones are unknown, precluding the development of targeted therapy for this HES variant. In this study, we used whole genome arrays to examine gene expression in the CD3−CD4+ T cells and found that 850 genes were differentially regulated during chronic disease compared with CD3+CD4+ T cells from healthy donors. Changes in the expression of 349 genes were altered in association with the clinical progression from chronic L-HES to T lymphoma in 1 patient, with 87 of 349 genes representing further changes in genes whose expression was altered in all chronic disease patients (87 of 850). Array analysis after CD2/CD28-mediated activation revealed that the major gene expression changes observed in the CD3−CD4+ T cells do not reflect activation induced alterations but rather pathways involved in T-cell homeostasis, including transforming growth factor-β signaling, apoptosis, and T-cell maturation, signaling, and migration. Examination of microRNA expression in the CD3−CD4+ T cells from patients with chronic disease identified 23 microRNAs that changed significantly, among which miR-125a further decreased in association with one patient's evolution to T lymphoma.
Introduction
Patients with lymphocytic variant hypereosinophilic syndrome (L-HES) are distinguished by the presence of abnormal T-cell populations (CD3−CD4+, CD3+CD4+, CD3+CD8+, or CD3+CD4−CD8−) that are frequently monoclonal.1 These clonal T cells secrete various combinations of interleukin-4 (IL-4), IL-5, and IL-13, resulting in hypereosinophilia and, in many cases, increased serum IgE levels.2,,,–6 Some patients with L-HES eventually develop peripheral T lymphoma2,4,7 with detection of an abnormal karyotype3,8,9 and resistance to apoptosis10 observed at preneoplastic disease stages. Despite this knowledge, T cell–mediated HES remains a heterogeneous group of diseases lacking definition of the molecular mechanisms underlying the persistence and expansion of the T-cell clone during chronic disease as well as the generation of increasingly abnormal subclones leading to T lymphoma. This contrasts with the discovery of the disease-inducing FIP1L1/PDGFRA fusion gene in HES patients with features of myeloproliferative disease, who are now treated with and remarkably responsive to the tyrosine kinase inhibitor imatinib mesylate.11
The goal of this study was to establish a molecular profile for CD3−CD4+ T cell–mediated L-HES by comparing gene expression (both mRNA and microRNA) in the abnormal T cells relative to normal CD3+CD4+ T cells. These analyses established a comprehensive immunophenotype/genotype that reflects the cells' Th2 nature as well as specific characteristics of polarization, signaling, and function. In patients with chronic disease, significant changes in gene expression were detected in critical growth control pathways of potential clinical relevance, including the IL-25 receptor and genes from the transforming growth factor-β (TGF-β) superfamily. Our previous studies found that the CD3−CD4+ T cells are dependent on exogenous T-cell receptor (TCR/CD3)–independent activation signals for Th2 cytokine expression.12 Assessment of gene expression changes associated with CD2/CD28-mediated costimulation revealed that the molecular alterations found in quiescent T cells associated with chronic disease did not simply reflect activation-associated changes in the abnormal T cells. Finally, we explored molecular changes linked with the outgrowth of a 6q-deleted subclone as one patient developed T lymphoma8 and found that approximately one-third of these genes were also altered in patients during chronic disease, suggesting that they may be of particular interest in terms of conferring a selective survival and growth advantage to the CD3−CD4+ T cells.
Methods
Detailed methods are provided in supplemental data (available on the Blood website; see the Supplemental Materials link at the top of the online article).
Patients
Patients with hypereosinophilic syndrome were selected for cohort inclusion based on the presence of a monoclonal CD3−CD4+ T-cell population in their peripheral blood. The clinical characteristics of all patients analyzed are summarized in supplemental Table 1. Informed consent was obtained from all patients in accordance with the Declaration of Helsinki, and this study was approved by the ethics committees at Institut Jules Bordet and Hôpital Erasme.
Cell purification
Circulating leukocytes were obtained from the peripheral blood of patients and healthy donors (controls) by venipuncture or cytapheresis, and peripheral blood mononuclear cells (PBMCs) were isolated and frozen as previously described.12 PBMCs were thawed and resuspended in X-vivo-20 medium (Lonza Braine SA). Patient CD3−CD4+ and donor CD3+CD4+ T cells were isolated by negative selection using magnetic beads (Miltenyi Biotec).8 The isolated cell populations were checked for purity by flow cytometry and were consistently more than 95% pure CD3−CD4+ (patients) or more than 90% pure CD3+CD4+ (controls). Thawed and purified T cells were incubated in X-vivo-20 at 37°C/5% CO2 for 18 hours to eliminate dying cells before RNA extraction. For the costimulation experiments, purified CD3−CD4+ T cells were cultured for 18 hours in X-vivo-20 supplemented with rhIL-2 (100 U/mL) and anti-CD28 (CLB-CD28/1; 1 μg/mL) plus 2 anti-CD2 antibodies (CLB-T11.1/1 and CLB-T11.2/1; 5 μg/mL each; Sanquin). In the IL-25 experiments, purified CD3−CD4+ (patient) and CD3+CD4+ (patient and control) T cells were stimulated for 48 hours with phorbol ester (10 ng/mL) and anti-CD28 in the absence or presence of increasing concentrations of rhIL-25 (R&D Systems).
RNA extraction and gene expression microarray procedures
RNA was extracted by the single-step method of isolation using Trizol (Invitrogen). RNA quantity and quality were assessed using a NanoDrop spectrophotometer (Thermo Fisher Scientific) and a Bioanalyzer (Agilent); 1.5 μg of total RNA was labeled following the manufacturer's protocols for probe preparation and hybridization (Affymetrix); 15 μg of cRNA was hybridized onto a U133 Plus 2.0 GeneChip.
Statistical analysis of microarray data
Raw data were analyzed using the SScoreBatch13,14 function from the SScore package (Version 1.5.1) in the R statistical environment (Version 2.3.0; http://www.r-project.org/; http://www.bioconductor.org). To identify genes consistently deregulated in the 3 patients relative to the 4 controls, we selected genes where each patient's P value (generated by S-score algorithm and Z transformation of mean S-score values) was inferior to .05 in comparison with individual controls (supplemental text and supplemental Table 2). A further filter was applied to probe sets of low significance. Similar analyses were performed to determine the genes deregulated during P1's evolution from chronic disease to T lymphoma (supplemental Table 3).
Flow cytometry
Flow cytometric analysis was performed by labeling 2 to 5 × 105 cells with 5 μL fluorescein isothiocyanate–conjugated anti-CD45RO, 5 μL peridinin chlorophyll protein-conjugated anti-CD3, 5 μL allophycocyanin-conjugated anti-CD4, and 5 μL of the phycoerythrin-conjugated test antibody (BD Biosciences; supplemental Table 4) in 50 μL X-vivo-20. A total of 10 000 viable cells were acquired on a FACS Calibur (BD Biosciences). Measurement of cytokine concentrations in culture supernatants was performed using BD Cytometric Bead Array Flex Sets.
Real-time quantitative RT-PCR
Total RNA (200 ng to 1 μg) was reverse-transcribed with random hexanucleotides using the SuperScript III First-Strand Synthesis System (Invitrogen) and standard protocols. Primers specific for the MAP3K8, RUNX1, RUNX2, DIABLO, TGFBR1, TGFBR2, TGFBR3, KIT, SMAD5, SMAD7, NOG, ACVR2A, and CYSLTR1 genes were purchased from QIAGEN (QuantiTect Primer Assays). Primers for ABL (endogenous control) were kindly provided by Dr J.-L. Vaerman. Quantitative reverse-transcribed polymerase chain reaction (RT-PCR) was performed on a Roche LightCycler 480 (Roche Applied Science). Analyses (supplemental Table 5) were performed using LightCycler Basic software, Version 1.5 (Roche).
MicroRNA quantification
Quantification of mature microRNAs was achieved using stem-loop-mediated RT-PCR with the TaqMan microRNA assay-early access kit or with individual microRNA assay mixes using the manufacturer's protocols (Applied Biosystems; supplemental Table 6). Standard real-time PCR was performed on an ABI Prism 7900HT (Applied Biosystems). Putative microRNA target genes were predicted using MiRanda algorithm-associated MirBase software (http://microrna.sanger.ac.uk).
Results
Comprehensive gene expression analysis of CD3−CD4+ T cells from L-HES patients
We compared the gene expression profiles of clonal CD3−CD4+ T cells isolated from L-HES patients (P1-P3; supplemental Table 1) during chronic disease with CD3+CD4+ T cells from controls. We also evaluated changes in gene expression associated with CD2/CD28 activation of their CD3−CD4+ T cells in vitro, an antibody combination targeting costimulatory receptors previously shown to mediate their Th2 cytokine production and proliferation.12 We further analyzed changes in gene expression linked with P1's clinical progression8,15 by assessing CD3−CD4+ T cells at diagnosis (yr 0), yr +4 (both premalignant stages of chronic L-HES), and yr +6 (concurrent with T lymphoma diagnosis). After comprehensive and stringent statistical analyses, we detected 850 genes (1397 probe sets) that were differentially regulated in all 3 patients CD3−CD4+ T cells compared with control CD3+CD4+ T cells (Figure 1; supplemental Table 2), 312 genes (411 probe sets) that were altered in all 3 patients CD3−CD4+ T cells after CD2/CD28 costimulation (supplemental Table 2), and 349 genes (450 probe sets) whose expression was altered in concert with P1's malignant evolution (supplemental Table 3). The original data for all 54 675 probes from each array are provided at Gene Expression Omnibus, http://www.ncbi.nlm.nih.gov/geo/, under accession number GSE12079.

Venn diagram of altered gene probe sets in the L-HES patients (P1-P3) vs controls. Significant changes in the expression of gene probe sets were based on a P < .05 after Z transformation of the mean S-score values obtained from all possible 2-chip comparisons between triplicates of P1-yr 0 or duplicates of P2-yr 0 and P3-yr 0 and individual arrays from 4 controls (supplemental data). The number of altered probe sets is shown, and the individual genes are listed in supplemental Table 2. *Among the 1469 commonly changed probe sets detected in the comparison of P1 to P3 vs controls, only 1397 probe sets passed a further filter that was applied to probe sets with a P > .01 and/or with fold change < 2 for at least 1 patient.
Venn diagram of altered gene probe sets in the L-HES patients (P1-P3) vs controls. Significant changes in the expression of gene probe sets were based on a P < .05 after Z transformation of the mean S-score values obtained from all possible 2-chip comparisons between triplicates of P1-yr 0 or duplicates of P2-yr 0 and P3-yr 0 and individual arrays from 4 controls (supplemental data). The number of altered probe sets is shown, and the individual genes are listed in supplemental Table 2. *Among the 1469 commonly changed probe sets detected in the comparison of P1 to P3 vs controls, only 1397 probe sets passed a further filter that was applied to probe sets with a P > .01 and/or with fold change < 2 for at least 1 patient.
Gene expression changes in CD3−CD4+ T cells from L-HES patients compared with CD3+CD4+ T cells from controls
Immunophenotype/genotype.
Based on the microarray data, we compiled a comprehensive phenotype/genotype of the CD3−CD4+ cells that extends previous characterizations of L-HES (Figure 2, supplemental Table 7). Prior studies of surface receptors on the CD3−CD4+ T cells2,5,9,10,12,15,–17 validated parallel changes observed in this study, including reduced or lost CD3 (CD3γ,CD3ζ), CD7, CD27 (TNFRSF7), and CD69 mRNA transcripts and increased CD5, CD95 (FAS) and HLA class II antigen mRNA transcripts. We determined whether altered mRNA expression corresponded to increased surface protein expression for numerous previously unexplored immunophenotypic markers using flow cytometry (supplemental Table 4) and an enlarged patient cohort (P1-P7; supplemental Table 1). Some gene expression changes can be attributed to the clonal Th2 nature of the patient's CD3−CD4+ T cells compared with the heterogeneous CD3+CD4+ T-cell population from controls.18 These include down-regulation of the Th1 genes BTLA, CCL5, IL-18R, NOTCH2, JUN, SLAMF7, and integrin α6 (ITGA6) and up-regulation of the established Th2 genes IL4R, CCR3, CCR8, GATA3, CRTH2 (CD294, GPR44), and IL17RB in the abnormal T cells.

Heat map of selected gene alterations detected in the comparison of L-HES patients (P1-P3) vs controls. A total of 198 of the commonly altered genes detected in all 3 patients were selected from supplemental Table 2 based on their functional relevance (one probe set/gene is shown and represents the greatest absolute fold change in patients relative to controls). The genes are classified in functional groups and listed in numerical order based on fold change with the same order and group numbers maintained in the heat map. Each column in the heat map represents the expression from an individual gene chip and includes the 4 healthy controls (C1-C4), triplicates of P1-yr 0 (a-c), and duplicates of P2 and P3 (a-b). The clustering and the heat map were generated using R 2.5.1. The dendrogram was derived from a group of selected genes using the hierarchical clustering method and shows the relatedness of gene expression patterns in the L-HES patients relative to the controls.
Heat map of selected gene alterations detected in the comparison of L-HES patients (P1-P3) vs controls. A total of 198 of the commonly altered genes detected in all 3 patients were selected from supplemental Table 2 based on their functional relevance (one probe set/gene is shown and represents the greatest absolute fold change in patients relative to controls). The genes are classified in functional groups and listed in numerical order based on fold change with the same order and group numbers maintained in the heat map. Each column in the heat map represents the expression from an individual gene chip and includes the 4 healthy controls (C1-C4), triplicates of P1-yr 0 (a-c), and duplicates of P2 and P3 (a-b). The clustering and the heat map were generated using R 2.5.1. The dendrogram was derived from a group of selected genes using the hierarchical clustering method and shows the relatedness of gene expression patterns in the L-HES patients relative to the controls.
IL-17RB, the receptor for IL-25 (IL17E), is expressed on memory Th2 cells19 and as recently shown also on human CD14+ cells.20 IL-25 induces Th2 inflammation in mice and, when bound to human Th2 cells, enhances Th2 cytokine production in response to T cell–stimulating agents.19 In this study, we detected increased expression of the IL17RB gene not only in all patients CD3−CD4+ T cells during chronic L-HES (Figure 2) but also observed further increases on P1's T lymphoma cells (Table 1) and after CD2/CD28 costimulation of P1 to P3's CD3−CD4+ T cells (Table 2). Flow cytometry detected a significantly higher proportion of P3's CD3−CD4+CD45RO+ T cells expressing membrane IL-17RB compared with CD3+CD4+CD45RO+ or CD45RO− T cells from controls (Figure 3A). P3's CD3−CD4+ T cells cultured with rhIL-25 induced a dose-response increase in the production of IL-5 and IL-13 but not interferon-γ and enhanced their proliferation in response to phorbol ester/anti-CD28 (Figure 3B). The further up-regulation of IL17RB mRNA observed on P1's T lymphoma cells and after CD2/CD28 costimulation of CD3−CD4+ T cells from chronic disease suggests that eosinophil-produced IL-25 may facilitate sustained and preferential expansion of the abnormal T-cell clone. Thus, increased IL17RB expression may provide a selective advantage to the CD3−CD4+ T-cell clone (or a subclone), thereby contributing to the maintenance of chronic disease and malignant progression.
Gene expression changes detected in CD3−CD4+ T cells from chronic L-HES patients relative to controls and in association with P1's evolution to T lymphoma

aFold change comparing the mean expression of duplicate arrays from P1, P2, and P3 with the mean expression from 4 controls; data are from supplemental Table 2.
bConsistently up-regulated genes are highlighted in dark gray; and consistently down-regulated genes in light gray.
cFold change comparing the mean expression of triplicate arrays from P1-yr 0 and P1-yr +6; data are from supplemental Table 3.
Changes in the expression of immune response genes associated with anti-CD2/CD28 activation of the CD3−CD4+ T cells from P1-P3
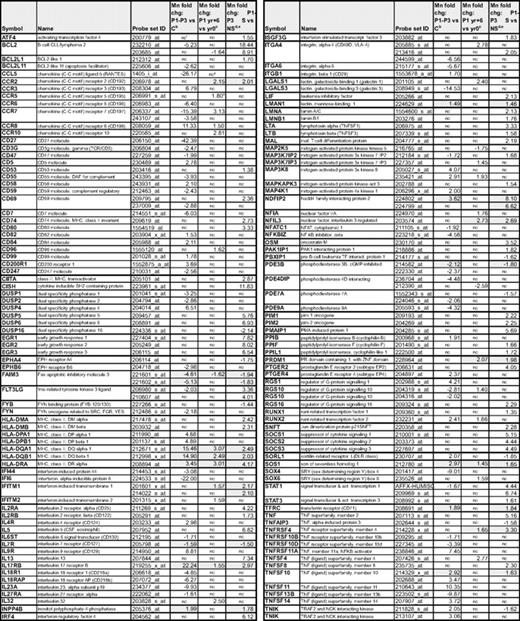
aOne probe (greatest fold change) is shown for each gene, except where differences were detected in individual probes to the same gene.
bFold change comparing the mean expression of duplicate arrays from P1, P2, and P3 nonstimulated with the mean expression from 4 controls; data are from supplemental Table 2.
cFold changes detected in the mean expression from triplicate arrays of P1 nonstimulated CD3−CD4+ T cells from yr +6 compared with yr 0; data are from supplemental Table 3.
dFold change comparing the mean expression of duplicate arrays from stimulated (S) vs nonstimulated (NS) CD3−CD4+ T cells from P1 to P3; data are from supplemental Table 2.
eGenes whose expression is similarly altered both after activation and in L-HES patient cells (either chronic disease or T lymphoma) are highlighted in gray.
fNC indicates no change.
gIn P1, CCL5 levels are decreased yr 0 vs C and yr +4 vs yr 0 but then increase in yr +6 vs yr 4 (supplemental Table 3).

IL-17RB (IL-25 receptor) and cytokine expression by L-HES CD3−CD4+ T cells. (A) Four-color immunofluorescent labeling of control and P3's PBMCs. The lymphocyte populations were gated on CD4 and CD3 positivity/negativity. (B) Purified CD3−CD4+ T cells from P3 were cultured for 48 hours with phorbol ester and anti-CD28 in the absence or presence of increasing concentrations of rhIL-25, and cytokine concentrations were determined using BD Cytometric Bead Array Flex Sets. A representative experiment is shown.
IL-17RB (IL-25 receptor) and cytokine expression by L-HES CD3−CD4+ T cells. (A) Four-color immunofluorescent labeling of control and P3's PBMCs. The lymphocyte populations were gated on CD4 and CD3 positivity/negativity. (B) Purified CD3−CD4+ T cells from P3 were cultured for 48 hours with phorbol ester and anti-CD28 in the absence or presence of increasing concentrations of rhIL-25, and cytokine concentrations were determined using BD Cytometric Bead Array Flex Sets. A representative experiment is shown.
Modulation of surface receptor expression is a common mechanism for controlling T cell–mediated immune responses that are characterized by cytokine production and proliferation. mRNA expression for several immunomodulatory genes other than the TCR/CD3 complex were altered in the patient's CD3−CD4+ T cells (Figure 2, supplemental Table 7), and some with apparent relevance include (1) decreases in the membrane complement regulatory proteins, which have been associated with autoimmune and inflammatory disease as well as the regulation of T-cell activation responses21 ; (2) decreased expression of genes involved in negatively regulating T-cell responses, including CTLA4 (CD152) and IL27RA, both shown to play critical roles in murine Th2 cell homeostasis22,23 and whose downmodulation potentially contributes to persistence and expansion of the CD3−CD4+ T-cell clone; and (3) increases in several immunoregulatory receptors, which perhaps provide a more pertinent characterization of costimulatory and regulatory pathways operating in the CD3−CD4+ T cells, including: (1) SLAMF5 (CD84), an inhibitory receptor for the high affinity IgE receptor24 ; (2) DCAL1 (CLECL1), a type II transmembrane C-type lectin-like protein expressed on dendritic cells and B cells (no previous reports of T-cell expression) and whose interaction with T cells has been shown to enhance their IL-4 production25 ; (3) CD99, a T-cell costimulatory receptor capable of fully activating cells stimulated with a suboptimal TCR/CD3 signal26 ; and (4) CD200R1, an inhibitory receptor regulating the activation threshold of inflammatory immune responses, which might also affect CD3−CD4+ T-cell activation.
The lack of TCR/CD3 expression on the abnormal T-cell surface dictates the potential loss of this important signaling pathway, although the CD3−CD4+ T cells do remain responsive to costimulatory signals.12 Some critical TCR/CD3 downstream signals were found decreased in unstimulated CD3−CD4+ T cells, including inhibitory receptors, PI3K-associated or family proteins, tyrosine and MAP3 kinases, and activation responsive transcription factors (supplemental Table 7). Interestingly, gene expression analysis of the abnormal T cells after CD2/CD28 costimulation revealed that relatively few of the gene expression changes detected in “quiescent” CD3−CD4+ T cells isolated from patient blood involved genes whose expression was affected by activation (Table 2, supplemental Table 2). Thus, induction of the Th2 cytokine genes IL5, IL13, and other immune response genes by costimulation in vitro apparently induces transient signals that may be elicited by a sustained stimulus present in local immune microenvironments in vivo.
G-protein–coupled receptors.
Numerous G-protein–coupled receptors were altered on the CD3−CD4+ T cells (Figure 2) and include 2 of particular significance. A 19-fold decrease in cysteinyl leukotriene receptor 1 (CYSLTR1) gene expression was observed in the CD3−CD4+ T cells with the greatest degree of down-modulation detected in P1-yr 0. Decreased CYSLTR1 expression was confirmed by quantitative RT-PCR (P1-P5, P7; Figure 4A, supplemental Table 5) but revealed a disparity in expression between the patients (ie, it is not expressed in P1 and low expression levels were detected in P2, P3, and P5). CYSLTR1 is normally expressed on myeloid cells, including eosinophils, and induced on CD4+ T cells by Th2 cytokines and TCR/CD3-mediated activation. The CYSLTR1 ligand, leukotriene D4, is produced by eosinophils and other myeloid cells and plays an active role both in cell survival and leukocyte recruitment to inflamed tissues.27 In contrast to the decreased expression observed on the CD3−CD4+ T cells, CYSLTR1 is significantly up-regulated and functional on CD4+ T cells from mice carrying an LAT gene mutation. These mice develop a Th2 lymphoproliferative disorder characterized by marked infiltration of CD3loCD4+ T cells in secondary lymphoid organs.28 Lack of CYSLTR1 on patients CD3−CD4+ T cells may render them less responsive to some eosinophil-derived survival signals and thereby contribute to the more indolent nature of L-HES.

Validation of changes in gene expression using quantitative RT-PCR and flow cytometry. (A-B) Fold change in the expression of selected genes measured by quantitative RT-PCR for (A) patients (P1-P3) relative to controls4 and (B) P1-yr +6 relative to P1-yr 0. P values were calculated based on 3 independent experiments using the Student t test and are indicated in the corresponding bar. (C) Histograms showing the surface expression of CD29 (ITGB1), CD49D (ITGA4), and CD62L (SELL) on control CD3+CD4+CD45RO+ T cells and P1-yr 0, P1-yr +4, and P1-yr +6 CD3−CD4+CD45RO+ T cells. Isotype controls (not shown) for each sample were set between 100 and 101.
Validation of changes in gene expression using quantitative RT-PCR and flow cytometry. (A-B) Fold change in the expression of selected genes measured by quantitative RT-PCR for (A) patients (P1-P3) relative to controls4 and (B) P1-yr +6 relative to P1-yr 0. P values were calculated based on 3 independent experiments using the Student t test and are indicated in the corresponding bar. (C) Histograms showing the surface expression of CD29 (ITGB1), CD49D (ITGA4), and CD62L (SELL) on control CD3+CD4+CD45RO+ T cells and P1-yr 0, P1-yr +4, and P1-yr +6 CD3−CD4+CD45RO+ T cells. Isotype controls (not shown) for each sample were set between 100 and 101.
The prostaglandin D2 receptor CRTH2, a G-protein–coupled receptor selectively expressed by Th2 cells, eosinophils, and basophils, is currently considered the most reliable marker for memory Th2 cells.29 Two CRTH2 (GPR44) gene probes revealed a 5- and 22-fold increase in expression in the patients abnormal T cells, which was confirmed by flow cytometry (supplemental Table 4). CRTH2 is involved in Th2 cell migration, GATA3 up-regulation, and induction of Th2 cytokine production.30 Our experiments found that GATA3 nuclear binding is up-regulated in patients CD3−CD4+ T cells only after CD2/CD28 costimulation31 ; however, the arrays detected a 3.6-fold increase in GATA3 transcripts in the patient's quiescent CD3−CD4+ T cells, which contrasts with the reported lack of differences in a microarray study of quiescent human Th1 and Th2 cells.32 Taken together, our data suggest that significant levels of GATA3 may be present in the cytoplasm waiting for activation signals that rapidly induce phosphorylation and nuclear translocation of this Th2 transcription factor, followed by cytokine gene up-regulation. The gene expression profiles of unstimulated versus CD2/CD28 costimulated CD3−CD4+ T cells clearly show that their Th2 cytokine expression is dependent on activation (Table 2). Together with our previous studies,12 these data suggest that, despite high CRTH2 and GATA3 expression levels, activating signals from local microenvironments in vivo are required to bring the circulating cells out of standby.
Apoptosis.
The clonal CD3−CD4+ T cells persist at relatively stable levels for many years in vivo, suggesting equilibrium between cell proliferation and apoptosis during chronic disease. The death domain containing tumor necrosis factor (TNF) superfamily plays critical roles in controlling the induction and progression of cell death, and the microarrays revealed altered expression of several TNF family member genes in the CD3−CD4+ T cells, including both proapoptotic genes and antiapoptotic (pro-proliferation) genes (Figure 2, supplemental Table 7); some were confirmed by quantitative RT-PCR (Figure 4A-B). Increased surface expression of FAS and a lack of CD27 in L-HES have been previously described2,10 and were reconfirmed in this cohort (supplemental Table 4). RANKL (TNFSF11; 10-fold increase) augments the costimulatory properties of antigen-presenting cells and thus could be important for CD3−CD4+ T cell activation in vivo. A 10-fold increase in RANKL mRNA was also detected in microarrays of CD4+ T cells from patients with Sezary syndrome.33 Interestingly, there was no further increase in RANKL expression associated with P1's progression to lymphoma or on costimulation. Further investigation into the functional implications of increased RANKL expression in L-HES is warranted given the ongoing development of humanized monoclonal antibodies for clinical use. The altered expression in specific subsets of genes involved in programmed cell death observed in this study suggests that there is a controlled balance that potentiates the increased survival and persistent expansion of the CD3−CD4+ T-cell clone.
T-cell homeostasis and the TGF-β family.
Altered expression among the TGF-β superfamily (TGF-β, activins, inhibins, growth differentiation factors, and bone morphogenetic proteins [BMP]) has been described for a variety of epithelial-derived solid tumors and hematologic malignancies.34 A microarray study revealed that TGF-β is the major signaling pathway that constitutively keeps human CD4+ T cells in a resting state.35 In this study, we detected numerous changes in TGF-β family gene expression in the L-HES CD3−CD4+ T cells during chronic disease, with a subset of these genes changing further during P1's evolution to T lymphoma (Figure 5, supplemental Table 7); in contrast, no additional changes were observed in the patients' abnormal T cells after CD2/CD28 costimulation. Decreased expression of the type I TGF-β receptor genes, TGF-βRI (TGFBR1) and ACVRIC, and the type II receptor gene TGF-βRII (TGFBR2) in the CD3−CD4+ T cells was confirmed by quantitative RT-PCR (Figure 4A). A previous study of CD4+ T-cell lines derived from T lymphoma patients found that decreased TGF-βRI and TGF-βRII expression was related to reduced responsiveness to TGF-β1-mediated growth inhibition,36 whereas microarrays of Sezary T cells detected TGFBR2 gene down-regulation.33

Schematic diagram of TGF-β signaling pathway. Altered expression in several TGF-β superfamily signaling genes was detected in L-HES patients CD3−CD4+ T cells. The mean fold changes detected in the microarrays for P1 to P3 relative to the controls are indicated in red (up-regulated genes) and green (down-regulated genes).
Schematic diagram of TGF-β signaling pathway. Altered expression in several TGF-β superfamily signaling genes was detected in L-HES patients CD3−CD4+ T cells. The mean fold changes detected in the microarrays for P1 to P3 relative to the controls are indicated in red (up-regulated genes) and green (down-regulated genes).
Studies have shown that a third TGF-β receptor, TGF-βRIII (TFGBR3; betaglycan) is frequently down-regulated in solid tumors34 in contrast to B chronic lymphocytic leukemia where its up-regulation has been reported.37 We detected increased TGFBR3 in the CD3−CD4+ T cells from patients with chronic disease. One study reported that corticosteroids can selectively stimulate TGF-βRIII expression in hepatic stellate cells38 ; and although corticosteroids are standard therapy for symptomatic L-HES patients, this treatment is probably not responsible for the TGFBR3 up-regulation observed because the fold changes for P2 and P3 (treated; supplemental Table 1) were lower than P1 (untreated).
TGF-βRIII binds all TGF-β isoforms and presents them to TGF-βRII, thereby initiating the recruitment and phosphorylation of TGF-βRI that leads to kinase activation. However, evidence indicates that TGF-βRIII also functions independently from TGF-β ligand presentation by working as a coreceptor with type 2 activin receptors (ACVR2A and ACVR2B, both genes increased in the CD3−CD4+ T cells). Activins and inhibins are structurally related members of the TGF-β superfamily that act as antagonists, with the former providing positive and the latter negative intracellular signals. High affinity binding of inhibin by TGF-βRIII is favored in cells coexpressing ACVR2A, thereby inhibiting the activin pathway. We also detected an increase in the BMP type I receptor gene, BMPRIA, which can interact with ACVR2A to bind BMPs. Finally, the noggin gene (NOG) was substantially decreased in the abnormal T cells and acts as an antagonist for the TGF-β superfamily members, BMP2 and BMP4, both of which play a role in early thymocyte differentiation.39 Altogether, these alterations in gene expression reflect a shift in the balance of TGF-β superfamily–dependent intracellular pathways in the CD3−CD4+ T cells, with uncontrolled signaling via the BMP pathway possibly disrupting normal homeostasis and favoring abnormal cell survival and growth.
This hypothesis is further substantiated by altered expression of Smad proteins, which transmit signals downstream from the TGF-β superfamily receptors. We observed increased receptor regulated SMAD5 gene expression together with decreases in the inhibitory SMAD7 gene, both confirmed by quantitative RT-PCR. Although their function in hematopoietic cells is not as well defined as Smad2, Smad3, and Smad4, Smad5 is involved in regulating BMP signaling whereas Smad7 negatively regulates receptor regulated Smad signaling and has been implicated in mature hematopoietic cell development.40 Receptor regulated Smad proteins specific for the BMP pathway, such as Smad5, interact with a variety of proteins, including Runx family transcription factors. The Runx genes have been shown to function as tumor suppressors in human cancer, although their overexpression in murine models revealed an oncogenic role in the development of hematopoietic tumors, including T lymphomas.41 Runx2 mediates cellular responses to signaling pathways hyperactive in tumors, including TGF-β family pathways, by forming coregulatory complexes with Smads and other coactivator and corepressor proteins to regulate gene transcription. Runx2, better known for its role in bone development and maintenance, was up-regulated in all patients and then again during P1's evolution to T lymphoma (Table 1). RUNX2 and RANKL (TNFSF11) are targets of transcriptional regulation by the vitamin D receptor (VDR; up-regulated in P1 yr +6) and were up-regulated in the CD3−CD4+ T cells from chronic disease. In addition, several target genes known to be induced by TGF-β were also decreased, including JUN, MYB, FLT3LG, and CXCR4 (the latter confirmed by flow cytometry). The clusterin gene (CLU), which interacts with TGF-βRII to modulate Smad signaling,42 was also significantly up-regulated in the abnormal T cells. Further investigation into the perturbations detected in the TGF-β superfamily signaling pathways and the role they play in the persistence of the CD3−CD4+ T-cell clone in L-HES are ongoing.
Gene expression changes in the CD3−CD4+ T cells associated with the evolution from chronic L-HES to T lymphoma
Cryopreserved blood samples from P1 spanning her 6-year progression from chronic disease to T lymphoma provided a very rare opportunity to assess changes in gene expression associated with malignant transformation in vivo. Our previous studies found that over time the initial CD3−CD4+ T-cell clone spawned subclones containing 2 independent 6q deletions (6q11-6q23.1 and 6q13-6q22.1) with progressive outgrowth of the 6q13q22.1-deleted subclone detected in 91% of the malignant T cells.2,8,15 Gene expression profiles of P1's CD3−CD4+ T cells at diagnosis (yr 0), during chronic disease (yr +4), and with T lymphoma (yr +6) revealed 349 genes (450 probe sets) that were differentially expressed in the malignant compared with the premalignant T cells (Table 3 and supplemental Table 3). Remarkably, approximately one-third of the probes (126 of 450), corresponding to 87 of 349 genes, were also initially altered in patients with chronic L-HES (Table 1), with the majority of these genes displaying stepwise alterations in expression (decreases or increases), first in all patients with chronic disease and then with P1's T lymphoma development. Progressive decreases were observed in several apoptosis genes, growth factors, and transcription factors along with progressive increases in surface receptors, signaling proteins, as well as additional growth factors and transcription factors. Among the genes whose expression changed progressively from chronic to malignant L-HES, only 6 genes (BCAT1, HLA-DQA1/2, HLA-DQB1, HLA-DRA, IL17RB, and SOS1) also increased and only the FAIM3 gene decreased after in vitro activation (Table 2). These data suggest that the genes whose expression is altered in the abnormal T cells from chronic and malignant L-HES reflect genuine changes in CD3−CD4+ T-cell physiology and not a transient response to external stimuli, such as signaling molecules, surface receptors, and cytokine production.
Selected gene expression changes in the CD3−CD4+ T cells during P1's evolution from chronic L-HES to lymphoma
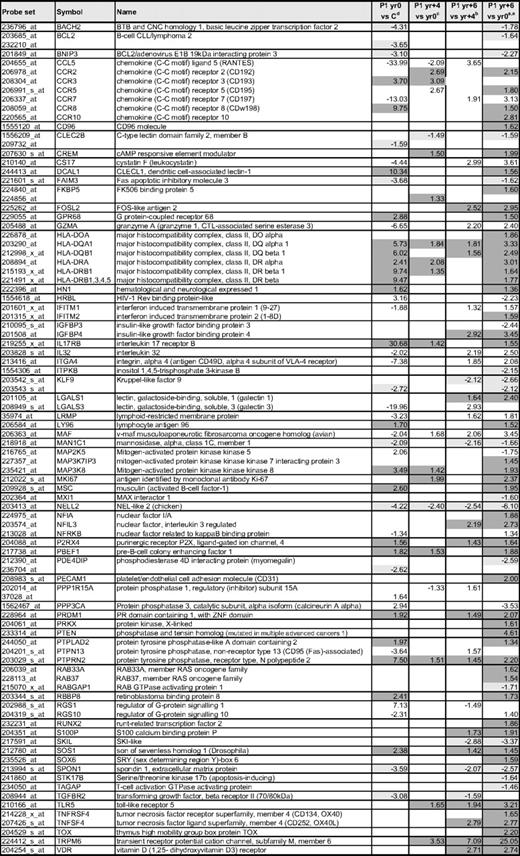
aFold changes detected in the mean expression from triplicate arrays of P1 CD3−CD4+ T cells from yr 0 compared with 4 controls; data are from supplemental Table 3. Progressively up-regulated genes are highlighted in dark gray and progressively down-regulated genes are highlighted in light gray.
bFold changes detected in the mean expression from triplicate arrays of P1 CD3−CD4+ T cells from yr +4 compared with yr 0; data are from supplemental Table 3.
cFold changes detected in the mean expression from triplicate arrays of P1 CD3−CD4+ T cells from yr +6 compared with yr +4; data are from supplemental Table 3.
dFold changes detected in the mean expression from triplicate arrays of P1 CD3−CD4+ T cells from yr +6 compared with yr 0; data are from supplemental Table 3.
Genes with potential relevance to malignant transformation include progressive increases in surface receptors, growth factors, transcription factors, and signaling proteins, as shown in Table 3. The limited number of genes whose expression decreases in the T lymphoma cells is distinguished by signaling proteins and transcription factors. One potentially relevant gene is the IL-9 receptor (IL9R), which was up-regulated on the CD3−CD4+ T cells from chronic L-HES; however, in contrast to its increased expression in mice overexpressing IL-9 that develop thymomas43 and some Hodgkin lymphomas, we did not observe further up-regulation on P1's T lymphoma cells. Overall, these alterations reflect progressive activation, altered signaling, and/or homing of the CD3−CD4+ T cells to specific sites and/or their adaptation to a specific microenvironment. The stepwise-modulated genes as well as those newly deregulated in the malignant T cells are of particular interest and relevance as potential therapeutic targets and new diagnostic markers.
T-cell trafficking and migration.
Leukocyte migration is mediated by a network of trafficking receptors expressed both on lymphoid and nonlymphoid tissues such that specific combinations of these adhesion and chemoattractant molecules act as traffic signals for directing extravasion and migration.44 Trafficking genes play distinct roles as leukocytes migrate through blood vessels. The initial step is mediated by selectins; and although we did not detect statistically significant changes in SELL (L-selectin; CD62L) gene expression, flow cytometry revealed surface receptor up-regulation on the CD3−CD4+ T cells, which continued to increase as P1 progressed to T lymphoma (Figure 4C). Rolling over endothelial cells exposes leukocytes to chemokines, which in turn provoke conformational changes in integrins that increase their affinity. The α4β1 integrin (VLA-4) is composed of 2 subunits, CD49D (α4; ITGA4) and CD29 (β1; ITGB1), both required for VLA-4 surface expression. Down-regulation of CD49D in association with a slight increase in CD29 was observed in the CD3−CD4+ T cells during chronic disease (Figures 2,4C and supplemental Tables 2,4). CD49D was reexpressed in concert with increased VLA-4 surface expression as P1 developed enlarged lymph nodes and progressed to T lymphoma (Table 1, Figure 4C).
Changes in other trafficking receptor genes were detected both in chronic disease and during the evolution to T lymphoma. Down-regulation of CXCR4, CXCR6, CCR6, and CCR7 was detected in patients during chronic disease with some CCR7 expression returning as P1 progressed to T lymphoma (Tables 1, 3). Increases in CCR3, ICAM3, LFA-3, CD82, and CD99 were observed in all patients with up-regulation of CCR2 in P1-yr +4 and CCR5, CCR10, CD96, and PECAM1 in P1-yr +6. CCR8 expression levels increased stepwise, 10-fold in chronic patients and a further 1.5-fold in P1-yr +6. CCR8 binds CCL1, which like CCL17 can be induced in bronchial epithelial cells by the Th2 cytokines IL-4 and IL-13.45 Although we have previously shown that serum CCL17 levels are extremely high in patients with L-HES,16 serum CCL1 levels and the functional role of CCR8 on CD3−CD4+ T cells remain unknown. The altered expression of trafficking receptors and ligands observed on the CD3−CD4+ T cells probably directs their movement to specific sites during premalignant and malignant L-HES disease,46 exposing the cells to external activation signals and/or costimulatory cells present locally.
Differential microRNA expression in the CD3−CD4+ T cells
MicroRNAs are endogenously expressed noncoding RNAs that regulate gene expression via mRNA degradation, mRNA destabilization, or translation inhibition. There is growing evidence that deregulated microRNA expression contributes to oncogenesis with an increasing number of identified microRNAs targeting genes involved in immune development, proliferation, and apoptosis.47 We extended the molecular profile of L-HES using quantitative RT-PCR to quantify changes in mature microRNA expression. Initially, we compared the expression of 156 microRNAs in CD3−CD4+ T cells from P1-yr.6 with control CD3+CD4+ T cells (supplemental Table 6). Thirty-eight microRNAs that decreased or increased greater than 2-fold in 2 independent experiments were selected for further analysis in CD3−CD4+ T cells from 6 chronic L-HES patients (P1-P5, P7) and CD3+CD4+ T cells from the same 4 controls. Using the nonpaired Student t test, we identified 23 microRNAs that were differentially expressed in the abnormal T cells (Table 4). The majority (19 of 23) of the selected microRNAs were down-regulated with increases found for only 4 microRNAs.
microRNAs that are differentially expressed in the CD3−CD4+ T cells from L-HES patients compared with CD3+CD4+ T cells from controls
| miRNA name | Patients vs controls | Chromosomal location‡ | |
|---|---|---|---|
| P* | Fold change† | ||
| hsa-let-7b | .032 | 3.2 | 22q13.31 |
| hsa-miR-26a | .019 | −2.3 | 3p22.3 |
| hsa-miR-31 | .004 | −111.4 | 9p21.3 |
| hsa-miR-95 | .025 | −2.6 | 4p16.1 |
| hsa-miR-99a | .011 | −60.9 | 21q21.1 |
| hsa-miR-100 | .010 | −57.6 | 11q24.1 |
| hsa-miR-126 | .030 | −9.1 | 9q34.3 |
| hsa-miR-130a | .034 | −6.2 | 11q12.1 |
| hsa-miR-135b | .011 | −11.8 | 1q32.1 |
| hsa-miR-135a | .008 | −10.9 | 3p21.1 |
| hsa-miR-151 | .019 | −12.1 | 8q24.3 |
| hsa-miR-181a | .010 | −34.6 | 1q31.3 |
| hsa-miR-181b | .010 | −19.3 | 1q31.3 |
| hsa-miR-193a | .017 | −4.6 | 17q11.2 |
| hsa-miR-213 | .011 | −78.8 | 1q31.3 |
| hsa-miR-215 | .019 | −3.1 | 1q41 |
| hsa-miR-221 | .010 | 3.4 | Xp11.3 |
| hsa-miR-222 | .010 | 3.7 | Xp11.3 |
| hsa-miR-335 | .010 | −8.0 | 7q32.2 |
| hsa-miR-340 | .019 | −4.9 | 5q35.3 |
| miRNA name | Patients vs controls | Chromosomal location‡ | |
|---|---|---|---|
| P* | Fold change† | ||
| hsa-let-7b | .032 | 3.2 | 22q13.31 |
| hsa-miR-26a | .019 | −2.3 | 3p22.3 |
| hsa-miR-31 | .004 | −111.4 | 9p21.3 |
| hsa-miR-95 | .025 | −2.6 | 4p16.1 |
| hsa-miR-99a | .011 | −60.9 | 21q21.1 |
| hsa-miR-100 | .010 | −57.6 | 11q24.1 |
| hsa-miR-126 | .030 | −9.1 | 9q34.3 |
| hsa-miR-130a | .034 | −6.2 | 11q12.1 |
| hsa-miR-135b | .011 | −11.8 | 1q32.1 |
| hsa-miR-135a | .008 | −10.9 | 3p21.1 |
| hsa-miR-151 | .019 | −12.1 | 8q24.3 |
| hsa-miR-181a | .010 | −34.6 | 1q31.3 |
| hsa-miR-181b | .010 | −19.3 | 1q31.3 |
| hsa-miR-193a | .017 | −4.6 | 17q11.2 |
| hsa-miR-213 | .011 | −78.8 | 1q31.3 |
| hsa-miR-215 | .019 | −3.1 | 1q41 |
| hsa-miR-221 | .010 | 3.4 | Xp11.3 |
| hsa-miR-222 | .010 | 3.7 | Xp11.3 |
| hsa-miR-335 | .010 | −8.0 | 7q32.2 |
| hsa-miR-340 | .019 | −4.9 | 5q35.3 |
P values were corrected using the false discovery rate calculation.
Fold change in the L-HES patients' CD3−CD4+ T cells (P1-P5 + P7) relative to controls (4).
Chromosomal locations were obtained from Ensembl.
One effect of the interaction between a microRNA and its target mRNA can be transcript cleavage and degradation. We searched for a correlation between global changes in mRNA and microRNA expression in the CD3−CD4+ T cells but did not observe a statistically significant correlation. We then used Ingenuity Pathways Analysis to assess the potential biologic importance of the predicted target genes as a group and determined that the best scored functional networks included the cell cycle, cell death, and hematologic system development and function. Individual microRNAs and their putative gene targets were generated using MirBase and included some of notable interest and potential relevance. The expression of 3 Th2 genes in the CD3−CD4+ T cells inversely paralleled several microRNAs predicted to target them, including increases in GATA3 with decreases in miR-10a, miR-95, and miR-130a, IL4R increases in concert with decreased miR-126 and miR-340, and increased CCR3 in parallel with decreased miR-181a, miR-181b, and miR-335. Genes whose mRNA expression changed in the abnormal T cells that were also predicted targets of 2 or 3 altered microRNAs included: IL18RAP (let7b, miR-221), CD99 (miR-31, miR-95, miR-135a), TRADD (miR-31, miR-125a), CD58 (miR-95, miR-135b), PPP3CA (miR-99a, miR-100), RANKL (TNFSF11; miR-126, miR-335), DMN3 (miR-126, miR-151), RGS1 (miR-130a, miR-335), and PRMT2 (miR-221, miR222).
Perhaps of greatest potential biologic significance were 3 genes whose expression increased in patients with chronic disease (RBBP8, CLU, and MAP3K8) with further increases associated with P1's evolution to T lymphoma (RBBP8 and MAP3K8) that were also predicted targets of 4 different down-regulated microRNAs. The retinoblastoma binding protein 8 gene (RBBP8), a predicted target of miR-31, miR-126, miR-130a, and miR-335, is thought to function as a tumor suppressor in conjunction with the transcriptional corepressor CTBP and BRCA1. Clusterin (CLU) is a calcium regulated protein whose expression has been associated with tumorigenesis and malignant progression, perhaps in part by modulating TGF-βRII signaling (Figure 5). The nuclear form is proapoptotic and the secretory form is antiapoptotic,48 with both forms involved in DNA repair and cell cycle regulation. Clusterin expression was significantly up-regulated in the CD3−CD4+ T cells in concert with the down-regulation of miR-99a, miR-100, miR-126, and miR-335. Thus, miR-126 and miR-335 potentially target both RBBP8 and CLU. A recent study of breast cancer found that miR-126 expression reduced tumor growth, whereas miR-335 suppressed lung and bone metastasis,49 with miR-335 loss leading to SOX4 and tenascin C (TNC) activation, which are both implicated in the acquisition of metastatic properties. We detected a 9-fold decrease in SOX4 (no change in TNC), which paralleled a 9-fold decrease in miR-335, suggesting that this microRNA targets other critical genes in T cells.
Many of the gene changes detected in patients relative to controls and then during P1's evolution to T lymphoma are involved in cell signaling. The MAP3K8 (Tpl2/Cot) oncogene expression increased stepwise first in patients during the chronic disease phase and again during P1's evolution to T lymphoma. The MAP3K8 gene is a predicted target of miR-135a, miR-135b, miR-181a, and miR-181b, which were all decreased in the CD3−CD4+ T cells. Studies have shown that decreased miR-181b expression in B chronic lymphocytic leukemia patients is associated with up-regulation of the TCL1 oncogene.50 The MAP3K8 gene is of particular interest because studies have shown that it is differentially regulated in hematopoietic cells and plays a role in tumor development. Overexpression and truncation of MAP3K8 lead to the activation of several T-cell signaling pathways and have been associated with large granular lymphocyte proliferative disorders.51 miR-181a also positively modulates TCR/CD3 sensitivity and affinity by suppressing phosphatases involved in negatively regulating TCR/CD3 signaling.52 The miR-181 family is involved in controlling hematopoietic cell differentiation and maturation with miR-181a levels fluctuating during thymopoiesis and its repression shown to diminish T-cell sensitivity in both primed and stimulated naive T cells.53 MiR-181a has also been shown to inhibit CD69, BCL2, and TCRα gene transcription.52 Intriguingly, our data revealed a substantial decrease in miR-181a and miR-181b associated with low CD69, BCL2, and TCR/CD3 expression levels in the CD3−CD4+ T cells, suggesting that the complex interactions between the TCR/CD3 signaling genes and the miR-181 family require further analysis. Experiments designed to approach the functional relevance of decreased expression of miR-135 family members, about which little is known, were accomplished by transfecting miR-135a and miR-135b mimics together in the CD4+ Jurkat T-cell line. These preliminary data indicate that the miR-135 mimics decrease MAP3K8 (−2.6-fold) and SMAD5 (−2.3-fold) expression compared with irrelevant sequence controls (data not shown).
Changes in microRNA expression during P1's clinical evolution
We also quantified expression of the same 38 microRNAs in association with P1's evolution to T lymphoma and found that only miR-125a changed significantly. miR-125a levels were 5.7-fold lower (P = .059) in the L-HES patient cohort relative to controls; and as P1 evolved to T lymphoma, miR-125a expression progressively decreased with an additional 2.8-fold drop (P = .003) detected at yr +6. Predicted gene targets of miR-125a were generated using MirBase and compared with the mRNA expression profiles of P1's CD3−CD4+ T cells (supplemental Table 6). The up-regulated target genes included another gene involved in signaling, PTPRN2, which is a member of the receptor-type protein tyrosine phosphatase family. PTPRN2 expression increased in parallel with the progressive decrease of miR-125a expression in the CD3−CD4+ T cells from chronic and malignant disease. PTPRN2 (IA-2β) is a pancreatic β-cell autoantigen for type 1 diabetes; and although its function is largely unknown, our studies suggest its role in L-HES warrants further investigation. A second miR-125a target gene, the Abelson helper integration site 1 (AHI1), was significantly up-regulated in the latter stages of P1's evolution to T lymphoma. AHI1 has been implicated in the development of T- and B-cell malignancies with increased expression detected in CD4+CD7− T cells from Sezary syndrome patients.54 Although the function of miR-125a remains unknown, its homolog miR-125b has been shown to posttrancriptionally target TNF-α and decrease cell proliferation.55 The stepwise down-regulation of miR-125a detected in L-HES disease suggests a potential role for this microRNA in the persistence and progressive transformation of the CD3−CD4+ T cells.
Discussion
L-HES associated with a clonal population of CD3−CD4+ Th2 cells is a rare benign lymphoproliferative disorder that can progress to malignant T lymphoma after a prolonged period of chronic disease. During the chronic phase, patients generally seek medical attention because of cutaneous symptoms, including severe eczema, angioedema, and urticaria. The Th2 nature of the underlying T-cell disorder is responsible for the marked hypereosinophilia, which also leads to frequent patient follow-up by physicians and treatment well before the development of malignancy. However, the mechanisms underlying clonal T-cell persistence and transformation remain unknown, precluding the development of a targeted therapy capable of eradicating the abnormal T cells during the chronic disease phase and thereby short-circuiting malignant transformation.1,56 The abnormal T cells persist for many years in vivo during which their levels are frequently stable in conjunction with corticosteroid treatment. The CD3−CD4+ T cells may eventually become refractory to treatment in parallel with malignant progression, which currently leaves only allogeneic stem cell transplantation as possibly curative.57
Our earlier work identified a CD3−CD4+ T-cell population as the source of Th2 cytokines in a symptomatic L-HES patient.17 We further demonstrated that the CD3−CD4+ T-cell population in patients was monoclonal based on its T-cell receptor rearrangement2 and that loss of surface TCR/CD3 expression was the result of a defect in CD3γ gene transcripts.15 We detected multiple chromosomal aberrations within the clonal T-cell population and found that evolution to malignancy in vivo was associated with the emergence of a subclone characterized by a specific 6q deletion in one patient.8 The present global gene expression study was undertaken to identify, in an unbiased manner, the specific genes and cellular pathways involved in the complex interplay between persistence and control of the CD3−CD4+ T cells in chronic L-HES and during their progression to full-blown malignancy.
The microarray analysis of patients with chronic disease provides a detailed immunophenotype/genotype, confirming the Th2 nature of the abnormal T-cell clone and offering insight on activation pathways and their homing state. Comparison of gene expression profiles from patients CD3−CD4+ T cells during chronic L-HES versus CD3+CD4+ T cells from healthy controls, activated or not by CD2/CD28 costimulation, demonstrated that altered gene expression in the abnormal T-cell clone does not simply reflect an activated memory T-cell phenotype. In addition, these data confirm that other previously reported functional characteristics of the CD3−CD4+ T cells, such as Th2 cytokine production and altered surface receptor expression, occur on engagement of membrane costimulatory receptors. We further assessed the importance of increased IL-25 receptor (IL-17RB) expression on the CD3−CD4+ T cells given the expected significant in vivo exposure to eosinophil-derived IL-25 in L-HES patients. These data demonstrate that the CD3−CD4+ T-cell response to IL-25 is characterized by Th2 cytokine production and increased proliferation in vitro. Given the premalignant nature of the CD3−CD4+ T cells during chronic disease, our findings indicate that controlling eosinophil levels should be a therapeutic endpoint for these patients, even though their frequently isolated cutaneous manifestations may not appear to warrant systemic therapy. Taken together, our data suggest that the blood-derived CD3−CD4+ T cells are in a transient state of ingress and egress with tissue microenvironments where they receive the signals for aberrant cytokine production and expansion.
We also identified genes whose expression deviated from the normal pattern of checks and balances controlling T-cell signaling and survival and thereby maintaining homeostasis. One of the more intriguing findings is the apparent switch in TGF-β superfamily signaling from TGF-β/Activin-directed to BMP-directed gene expression (Figure 5). TGF-β has been extensively characterized for its immune suppressive functions and is known to play critical roles in controlling thymocyte development and limiting effector/memory T-cell responses. Activin A is produced by activated Th2 cells and plays a role in Th2-mediated responses of B cells and macrophages.58 BMPs were initially identified for their growth factor effects on bone formation but have since been shown to regulate neurogenesis and hematopoiesis during embryonic development; and although little is known about BMP-mediated control of mature T-cell responses, BMPs have been shown to play a role in T-cell differentiation in the thymus.39 Several studies have described aberrations in BMP signaling pathways in solid tumors, suggesting that survival and expansion of the CD3−CD4+ T-cell clone in L-HES could be in part the result of a switch from negative TGF-β regulation to positive BMP signaling, and the processes involved are currently under investigation.
The sequential analysis of P1's clinical evolution revealed that almost one-third of the genes whose expression changed in association with the development of T lymphoma were already abnormally expressed in L-HES patients during chronic disease. The majority of these genes were not altered in response to in vitro activation, further suggesting that they reflect inherent changes in CD3−CD4+ T-cell biology associated with transformation and deregulated growth. These genes include progressively deregulated oncogenes, transcription factors, and signaling genes. Together with the genes that were newly altered in P1's T lymphoma cells, this relatively small number of genes identifies critical players in chronic and malignant L-HES. Good examples are the 3 genes, RBBP8, MAP3K8, and PTPRN2, whose expression increased stepwise CD3−CD4+ T cells, first in chronic disease and then in association with T lymphoma. Furthermore, their increased expression paralleled decreases in microRNAs predicted to target them, and our preliminary miRNA transfection experiments lend credence to the functional consequences. For example, the MAP3K8 oncogene is a member of the serine/threonine protein kinase family that was identified by its transforming activity. Activated MAP3K8 induces the ERK1/2, JNK, NF-κB, and p38MAPK pathways, and a study has shown that it is constitutively activated in HTLV-I-transformed human CD4+ T-cell lines.59 The MAP3K8 gene therefore illustrates a gene deregulation (increased expression) detected in our patient cohort during chronic disease, which was further augmented in L-HES–associated T lymphoma and identified as a potential target of microRNAs shown to be downmodulated in the patients' cells.
Our objective in this study was to provide a global assessment of gene expression changes characteristic of the CD3−CD4+ T cells during chronic and malignant L-HES as a means of identifying the deregulated pathways that underlie their abnormal persistence and expansion in vivo. These data reveal important gene expression changes in receptors whose altered expression may contribute to the CD3−CD4+ T cell–modified responses to environmental stimuli as well as deviations in homeostatic growth control pathways whose perturbations may favor outgrowth of the abnormal T-cell clone. Preliminary functional experiments confirmed that the aberrant pathways identified in the CD3−CD4+ T cells warrant further in-depth exploration, and several specifically deregulated genes point to potential new drug targets and diagnostic markers.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Drs Johannes Huss-Marp, André Efira, and Bernard Kennes, who referred HES patients and/or provided samples for this study; Carole Equeter and Françoise Lallemand for help with the microarray hybridizations; and Floriane Andre for technical assistance.
This work was supported by grants from the Belgian Fund for Scientific Research (FNRS), Opération Télévie, Les Amis de l'Institut Bordet, Fondation Bekales, the International Brachet Stufting, Fondation Salus Sanguinis, and the European Union Framework Program 6 (LSHB-CT-2005-018680). K.W.-G. is a scientific collaborator of the FNRS-Télévie. M.R. was a fellow of the FNRS-Télévie. C.G. is a fellow of the FNRS-Télévie.
Authorship
Contribution: M.R. and K.W.-G. designed the research; M.R. and M.L. performed the research; M.R., M.L., C. Sibille, F.R., M.G., and K.W.-G. analyzed and interpreted the data; C. Sotiriou contributed vital analytical tools; C.G. and B.H.-K. performed the statistical analysis of microarray data; and M.R., F.R., and K.W.-G. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Karen Willard-Gallo, Molecular Immunology Unit, Institut Jules Bordet, Université Libre de Bruxelles, 121 Blvd de Waterloo, B1000 Brussels, Belgium; e-mail: kwillard@ulb.ac.be.
