

Abstract
Gene expression in cells is a dynamic process but is usually examined at a single time point. We used gene expression profiling over time to build temporal models of gene transcription after B-cell receptor (BCR) signaling in healthy and malignant B cells and chose this as a model since BCR cross-linking induces both cell proliferation and apoptosis, with increased apoptosis in chronic lymphocytic leukemia (CLL) compared to healthy B cells. To determine the basis for this, we examined the global temporal gene expression profile for BCR stimulation and developed a linear combination method to summarize the effect of BCR simulation over all the time points for all patients. Functional learning identified common early events in healthy B cells and CLL cells. Although healthy and malignant B cells share a common genetic pattern early after BCR signaling, a specific genetic program is engaged by the malignant cells at later time points after BCR stimulation. These findings identify the molecular basis for the different functional consequences of BCR cross-linking in healthy and malignant B cells. Analysis of gene expression profiling over time may be used to identify genes that might be rational targets to perturb these pathways.
Introduction
There is a steady state of transcription that differs in healthy and malignant cells and that can be captured by gene expression profiling. However, the transcriptional program downstream of a cellular stimulus is a dynamic temporal process. After cell stimulation, the concerted action of thousands of genes and their products leads to the programmed cell reaction. High throughput tools, including gene expression profiling, now enable us to examine changes in expression of genes after stimulation, but the challenge that remains is how to integrate these data. Although temporal clustering of expressed genes after intervention have shown finely structured patterns of transcription,1 organization of such gene expression data into meaningful biologic data has proven difficult.2
We chose to examine the temporal changes in gene expression after B-cell receptor (BCR)–mediated signaling to examine changes in transcription in healthy and malignant cells as a model of how to integrate such data. The BCR on mature B cells consists of an antigen-binding subunit, surface immunoglobulin (sIg), and a signaling subunit consisting of CD79a and CD79b. Cell-urface expression of the BCR is required for B-cell development, tolerance to self-antigens, and response to antigen. Cross-linking of the BCR initiates a tyrosine kinase cascade, and regulation of the BCR signaling pathway by factors, including the strength of the BCR-mediated signal, determines whether BCR engagement leads to activation, anergy, or cell death by apoptosis.3,4 The temporal genetic program after BCR stimulation has not previously been explored in primary human B cells and has been studied in murine cells only at 2 time points.5
Since BCR signaling also plays an important role in maintenance and proliferation of malignant B cells, we compared healthy peripheral-blood B cells and B-cell chronic lymphocytic leukemia (CLL) cells. There is increasing evidence that diseases such as CLL may be an antigen-driven process based on restriction of the repertory as well as shared antigen-binding motifs used by CLL B cells.6–8 An important prognostic factor in CLL is the presence or absence of somatic hypermutation of the Ig heavy and light chains, with those cases with unmutated immunoglobulin variable heavy chain (IgVH) genes (UM-CLL) having a more aggressive course compared to the more indolent IgVH mutated (M-CLL) cases.9,10 Regardless of the mutational status, CLL cases share a common basal gene expression profile related to antigen-stimulated mature B lymphocytes,11–13 although a number of genes were identified that discriminate mutated from unmutated cases. Most notable among these is zeta-associated protein of 70 kDa (ZAP70), a receptor-associated protein tyrosine kinase (PTK) expressed by T lymphocytes, natural killer (NK) cells,14 and a subpopulation of normal B cells. ZAP70 has increased expression in UM-CLL and has prognostic significance.15–18 ZAP70 appears to play a role in enhancing BCR signaling of more aggressive CLL19–21 and may play a role by modulating the strength of BCR-mediated signaling in the more aggressive nature of some cases of CLL.22
Here we demonstrate the early and later transcriptional consequences of BCR cross-linking and identify that there is a common structure of functional gene groups in murine and human B cells as well as in healthy and malignant B cells. In the malignant cells, differentially expressed genes can potentiallyinterfere with these functional groups, most notably in ways that influence cell proliferation and apoptosis.
Patients, materials, and methods
Subjects and B-cell isolation
Peripheral blood was obtained from 12 previously untreated patients diagnosed with B-CLL and 6 healthy blood donors. Written informed consent was obtained from all patients in accordance with the Declaration of Helsinki, and approval to obtain and study the samples was obtained from the Institutional Review Board at Dana-Farber Cancer Institute. Patients with indolent disease had stable disease over time, while patients with aggressive disease had a rapid clinical course; all these patients have since required treatment for their disease. IgVH gene mutation status and ZAP70 expression were evaluated for each patient.18 Cytogenetic abnormalities were identified by metaphase analysis and by fluorescence in situ hybridization (FISH) using a panel of probes.23
B cells were negatively selected from fresh blood samples using the Rosettesep B-cell enrichment cocktail (StemCell Technologies, Vancouver, BC, Canada) and isolated by density gradient centrifugation over Ficoll-Paque Plus (Pharmacia, Uppsala, Sweden). Quality of the selection was assessed by flow cytometry on a Cytomics FC500 System (Beckman-Coulter, Fullerton, CA) after CD5-PE/CD19-FITC staining (BD Biosciences, Palo Alto, CA) and ranged from 93% to 98% (median, 96%). Cells were serum starved for 4 hours at 37°C/5% CO2 in RPMI 1640 media supplemented with 2 nM L glutamine and 24 μg/mL gentamicin, as previously described.20
BCR stimulation and sample collection
B cells, at a density of 107cells/mL, were divided in 2 to control for any effect of ex vivo handling of cells over the time period of the experiment. One was used as unstimulated (US) control, and the other was stimulated (S) by goat F(ab')2 anti–human IgM-BIOT (Southern Biotechnology, Birmingham, AL) at 20 μg/mL and cross-linked by 20 μg/mL avidin (Sigma-Aldrich, St Louis, MO) on ice for 5 minutes, followed by incubation at 37°C for 15 minutes. The amount and duration of stimulation were determined in a pilot study evaluating the apoptosis response assessed by flow cytometry as described below under “Apoptosis assay” as well as protein tyrosine kinase phosphorylation immunoblotting (data not shown). After stimulation, cells were washed, resuspended in RPMI-1640 culture medium supplemented with 10% heat-inactivated FCS, 2 mM L-glutamine, and 24 μg/mL gentamicin, and cultured at 37°C in 5% CO2.
Four time points (60, 90, 210, and 390 minutes) after BCR stimulation were chosen to collect total RNA samples after a pilot study from 20 minutes to 24 hours after BCR engagement. The first US time point was considered as baseline. At each time point, 107 control US and S cells were washed and resuspended in 1 mL TRIzol (Invitrogen, Carlsbad, CA). Total RNA was purified using Promega columns (SV total isolation system; Promega, Madison, WI). RNA quality was assessed using the 2100 Agilent Bioanalyzer (Agilent, Palo Alto, CA). At these 4 time points and a later time point (16 hours), 107 control US and S cells were washed, pelleted, and lysed in ice-cold 1% Ipegal (NP-40; Sigma) lysis buffer for 20 minutes on ice. Cell lysates were clarified by centrifugation at 20 000 g for 15 minutes. Protein concentration was determined using the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA).
Apoptosis assay
Apoptosis response after BCR stimulation was evaluated by flow cytometry. Before BCR engagement and at several time points after stimulation, 0.4 × 106 US and S cells were isolated, washed in PBS, and stained with Annexin V FITC and propidium iodide (BD Biosciences). All analyses were made with an FC 500, and results were analyzed with RXP software (BD Biosciences). After a pilot study over 24 hours, the Overton subtraction of fluorescence (S-US) at 6 hours after the BCR engagement was calculated to evaluate the B-cell apoptosis response.
Microarray experiments
cRNA was prepared in accordance with the Affymetrix (Santa Clara, CA) protocol and hybridized to the HG-HU133 plus 2.0 microarray containing 54 675 probe sets. A pilot experiment including 3 patients (UM-CLL—apoptosis responder, UM-CLL—apoptosis low responder, M-CLL—median apoptosis responder) was conducted to select the best time points for analyzing functional transcription over 24 hours after BCR engagement. For both S and US cells, 8 time points—20 minutes, 30 minutes, 60 minutes, 90 minutes, 150 minutes, 210 minutes, 390 minutes, and 24 hours—were analyzed for each of these 3 patients, so that a total of 48 samples were included in the pilot study. After selecting the 4 best time points (60, 90, 210, and 390 minutes), we further generated 120 microarrays consisting of 4 time points for S and US cells in 15 additional cases (9 CLL patients and 6 healthy donors), so that a total of 12 CLL and 6 healthy donors were analyzed. Among a total of 168 samples, 144 samples were included in the further analyses, excluding the additional time points from the pilot series. Data were normalized with the invariant set method and the model-based expression index (MBEI) obtained by the PM-mm model using dChip software (dChip).24 We preprocessed the expression value by truncating the value to less than or equal to 1.
Time point selection
The pilot experiment consisting of 48 samples of 8 time points for S and US cells from 3 patients was analyzed to select the most important among the 6 time points to design the further experiment. We ran self-organizing map (SOM)25 using the log2 ratio of the S/US expression value in an unsupervised manner to examine if there were biologic time trends detected and selected 8 possible template patterns of gene expression over time. We obtained the list of differentially stimulated genes having at least 1.5 fold-changes from baseline comparing the 3 patients included in the pilot study. We considered the time trend patterns that appeared in these differentially stimulated genes as important time trend patterns. Fisher exact test26 comparing the number of the differentially stimulated genes and the gene list having the Spearman correlation greater than 0.8 with the 8 template patterns was performed for each patient, and 6 patterns were commonly found to have significant trends in differentially stimulated genes. The time points 60, 90, 210, and 390 minutes were selected that contributed to 5 significant time trend patterns (monotonously increasing or decreasing BCR-simulated effects, increasing in 60-90 minutes/60-210 minutes and decreasing in the later time points, and decreasing in 60-90 minutes and increasing in the later time points) to be further studied in 15 additional subjects.
Statistical analysis
Statistical analyses were performed by R software (http://www.r-project.org/), dChip,24 and GeneCluster 2.0 (http://www.broad.mit.edu/cancer/software/genecluster2/gc2.html). Hierarchical clustering analyses27 were performed by dChip software with 1-correlation as the distance metric and centroid linkage method. To perform the clustering for the samples, we further filtered genes to obtain fewer than 1000 genes. The variation across samples measured by standard deviation/mean of gene expression levels and a call of present is greater than or equal to 20% in the array were used as filtering criteria. This filtering step was found to be robust in terms of clustering results with respect to various filtering criteria. Three separate hierarchical clusterings were performed: (1) hierarchical clustering analysis for all 144 samples was performed with the 925 filtered probe set; (2) hierarchical clustering analyses of the 18 basal unstimulated gene expression was performed with 906 filtered probe sets; (3) hierarchical clustering for the temporal changes using a linear combination over time points to make a score for stimulated expressions over time as,
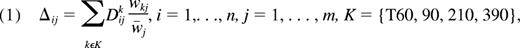
where i, j, and k are indicating the sample, gene, and time, respectively. We used the inverse of the standard error of samples for each gene j of time k as a weight wkj and

We performed the hierarchical clustering of 18 samples for this score Δ to examine the effect of the all time points with the 860 filtered probe sets.
Identification of marker gene lists of differentially expressed or stimulated genes in supervised analyses
Based on the 5688 filtered genes, marker gene lists were found by comparing 2 groups defined from the unsupervised hierarchical clustering, as follows: (1) marker gene list of the basal unstimulated gene expression (T60 US) used the compare sample function in dChip with P < .05 (t test) and fold change greater than or equal to 1.5, with 90% confidence interval for selecting significantly differentially expressed genes; (2) marker gene list of the temporal changes of BCR-stimulated effect across 4 time points. We first selected genes that had a median of P values from 4 t tests for testing the difference of Dk, k = T60, 90, 210, 390 and had P values testing the difference of Δ between 2 groups less than 0.1 (t test). Then we tested the null hypothesis; there are no differences between the distribution functions of (D1, D2, D3, D4) for 2 groups using Wei and Johnson's method.28 This procedure gives a locally best-linear combination of test statistics to summarize the stimulation effect over all time points without assuming any model of dependence among time points. We calculated the P value by computing the locally best combination of 4 t statistics and used permutation to compute the background noise. We obtained marker gene lists that had P ≤ .05.
Pathway analyses
We used Ingenuity Pathways Analysis software (Ingenuity Systems, Redwood City, CA) to identify potentially deregulated pathways within the cells by visualizing a relevant functional network of expressed genes at http://www.ingenuity.com/products/pathways_analysis.html.
Housekeeping gene selection
For every gene and every patient, we calculated the mean expression level across all 8 time points (4 S, 4 US). Within each patient, we marked every gene that is always within ± 5% of its mean value (specific to the patient). From that subset of genes, we calculated the mean expression level across all 8 time points and all patients for every gene. We marked every gene that is within ± 5% of the overall mean expression level for that gene in at least 4 patients.
Real-time quantitative PCR gene expression
We performed real-time quantitative PCR (RQ-PCR) validation for 15 subjects (12 CLL, 3 healthy) with an aliquot of total RNA previously used for microarray hybridization. First-strand cDNA was synthesized with an aliquot of 1 μg total RNA using oligo d(T) primers (Invitrogen, Carlsbad, CA) and SuperScript III RNase H reverse transcriptase (Invitrogen). Primer Express 2.0 was used to design specific primers (Invitrogen) and probes (Integrated DNA Technologies, Coralville, IA) for DUSP1, EGR2, and NFκB1 as genes of interest and RPS28 as a housekeeping gene. Each gene was cloned using TopoA vector (Invitrogen) to establish a dilution of each plasmid DNA. RQ-PCR was performed on ABI Prism 7000 (Applied Biosystems, Foster City, CA) using a final concentration of 100 nM probe, 400 nM primer, 1 × Quantitect Probe PCR kit (Qiagen, Valencia, CA) and 25 ng/μL cDNA in a 50-μL reaction volume using annealing and extension temperatures of 60° for 60 seconds. The ΔCT (threshold cycle) value of mean CT of the gene of interest using the standard curve method established by triplicate quantification of dilution of plasmid DNA of the gene of interest in the same plate was calculated. We then calculated the ΔCT of mean CT of the housekeeping gene in the same cDNA using the standard curve method established by quantification in triplicate of dilution of plasmid DNA of the housekeeping gene. Mean difference CT is always less than 0.5 CT. We calculated the ratio of RNA copy number quantity between each gene and the housekeeping gene. Finally, the delta (S-US) of the ratio of expression between the gene of interest and the housekeeping gene for each time point is used to calculate the expression.
Immunoblot analysis
Thirty micrograms of total protein was loaded onto 10% SDS–polyacrylamide gel for electrophoresis. The size-separated proteins were transferred to PVDF Hybond-P membrane (Amersham Biosciences, Piscataway, NJ) and were blotted with primary antibodies (R&D Systems, Minneapolis, MN; BD Biosciences). Secondary horseradish peroxidase–conjugated antibodies (Sigma-Aldrich) were detected using enhanced chemiluminescence (ECL; Amersham Biosciences) and autoradiography with hyperfilm ECL (Amersham Biosciences).
Results
Pilot study
We first performed an exploratory pilot study at the whole genome level in human cells at multiple time points over 24 hours. We stimulated the BCR pathway in 3 selected CLL B cells, as described in “Subjects, materials, and methods.” Total RNA was extracted from stimulated (S) and control unstimulated (US) cells at multiple time points after BCR cross-linking (baseline, 20, 30, 60, 90, 150, 210, 390 minutes, and 24 hours), and gene expression analysis was performed. The pilot experiment therefore consisted of 48 samples of baseline plus 8 subsequent time points for both the S and US cells and was analyzed using unsupervised self-organized maps to search for transcriptional time trends after stimulation to select the most important time points to design subsequent experiments. Based upon this pilot study, we identified that selection of 2 early time points (60 and 90 minutes), 1 intermediate time point (210 minutes), and 1 later time point (390 minute) captured the vast majority of genes constituting the BCR transcriptional program. We noted a change in gene expression profiles over time in the unstimulated cells, demonstrating the necessity to acquire control US gene expression for each selected time point for all future experiments. We analyzed the BCR transcriptional program in negatively selected fresh US and S B cells isolated from 12 CLL patients and 6 healthy donors at the 4 time points established for analysis so that a total of 144 HG-HU133 plus 2.0 microarrays containing 54 675 probe sets were analyzed.
Temporal gene expression: description
The first aim of the study was to unravel the temporal transcriptional program used by healthy and leukemic B cells. Before assessing this temporal process, we considered the basal expression data at the first unstimulated time point. We performed unsupervised learning of the baseline gene expression of the 12 CLL patients and 6 healthy donors. As expected,11,12 unsupervised hierarchical clustering of the samples distinguishes healthy B cells from CLL cells (Figure 1). Five hundred eight marker probe sets (273 overexpressed and 235 underexpressed in CLL vs healthy cells) were statistically differentially expressed (P = .05) in CLL patients compared to healthy donors. Because of redundancy within the Affymetrix probe sets, 426 genes (219 overexpressed and 207 underexpressed in CLL vs healthy cells) were statistically differentially expressed, among which 237 are known genes with Human Genome Organization (HUGO) defined gene symbols. Of note, overexpressed genes in CLL compared to healthy cells included ZAP70, SYK, and LCK.

Unsupervised hierarchical clustering of the basal gene expression discriminates CLL B cells from healthy B cells. Five hundred eight marker probe sets were statistically differentially expressed (P = .05) in CLL patients compared to healthy donors.
Unsupervised hierarchical clustering of the basal gene expression discriminates CLL B cells from healthy B cells. Five hundred eight marker probe sets were statistically differentially expressed (P = .05) in CLL patients compared to healthy donors.
Global gene expressions from the entire subject at each time point were then considered, with analysis of all 144 samples for the 18 subjects at the 4 selected time points. Data were preprocessed, and genes were filtered to identify those with a consistent pattern of expression among those subject to BCR stimulation across time. We examined the effect on gene expression of BCR simulation at each of the 4 time points by calculating the difference (D) of expression between the S and US samples for each patient: Dk = (X
Unsupervised hierarchical clustering analysis was performed with the 925 probe sets, which identified 3 main clusters (Figure 2). A first cluster merges together the healthy B-cells samples at all unstimulated time points and early stimulated (60, 90 minutes) time points. The second cluster merges the CLL samples at the early stimulated time points. The third cluster merges together the healthy B cells and the CLL samples at the intermediate and later time points. The BCR-stimulated expressions of these filtered genes drive the clustering process both on early and leukemic B cells, constituting a common core BCR gene expression signature.

Functional gene expression after BCR cross-linking. Each time point is shown as a column, with 4 time points for unstimulated and 4 time points for stimulated cells, for each of the 18 subjects (6 healthy and 12 CLL patients) for a total of 144 DNA chips. Nine hundred twenty-five of 54 675 probe sets satisfied the filtering criteria. Unsupervised hierarchical clustering analysis of functional gene expression after BCR cross-linking.
Functional gene expression after BCR cross-linking. Each time point is shown as a column, with 4 time points for unstimulated and 4 time points for stimulated cells, for each of the 18 subjects (6 healthy and 12 CLL patients) for a total of 144 DNA chips. Nine hundred twenty-five of 54 675 probe sets satisfied the filtering criteria. Unsupervised hierarchical clustering analysis of functional gene expression after BCR cross-linking.
Temporal gene expression: comparison
In view of this finding that a core BCR gene expression signature was shared by all the B cells, our next goal was to look for any leukemic specific alteration of this BCR temporal genetic program. We compared the temporal gene expression of each subject across time without any a priori categorization. For each subject, temporal expression(s) at each time point was normalized by the control US expression data, and the linear combination of the delta (S-US) of expression was then computed for each gene across time. Unsupervised clustering was then performed using the result of the temporal combination of each gene for each subject across time, as described. This unsupervised clustering revealed a differential temporal gene expression between healthy and leukemic cells and, of interest, also differentiated 2 patient clusters (Figure 3). The clinical and biologic characteristics of the patients showed that the temporal gene expression differentiated patients with a more aggressive clinical course (UM-CLL, ZAP70 positive, adverse cytogenetics aberration, progressive CLL) from those with indolent leukemia (M-CLL, ZAP70 negative, no adverse cytogenetic aberration, indolent clinical course). This temporal gene expression learning revealed a BCR transcriptional specific for the more aggressive leukemic cells.

Linear combination of marker probe sets. (A) Unsupervised linear combination of expression of the 18 subjects over 4 time points after BCR crosslinking. Eight hundred sixty of 54 675 probe sets satisfied the filtering criteria. (B) Probe sets differentially expressed over time between UM-CLL Zap+ B cells and M-CLL Zap− B cells. One hundred eighteen of 54 675 probe sets satisfied the filtering criteria. (C) Probe sets differentially expressed over time between healthy B cells and CLL patient marker genes. One hundred four of 54 675 probe sets satisfied the filtering criteria.
Linear combination of marker probe sets. (A) Unsupervised linear combination of expression of the 18 subjects over 4 time points after BCR crosslinking. Eight hundred sixty of 54 675 probe sets satisfied the filtering criteria. (B) Probe sets differentially expressed over time between UM-CLL Zap+ B cells and M-CLL Zap− B cells. One hundred eighteen of 54 675 probe sets satisfied the filtering criteria. (C) Probe sets differentially expressed over time between healthy B cells and CLL patient marker genes. One hundred four of 54 675 probe sets satisfied the filtering criteria.
Based upon this unsupervised clustering, we sought to identify the marker gene list differentially expressed between the different cell populations and identified 75 different known genes (104 marker probe sets) differentially expressed between healthy and leukemic cells as well as 99 different known genes (118 marker probe sets) differentially expressed between aggressive and indolent leukemic cells. These results suggest that in addition to a core BCR gene expression program shared by every B cell there is a temporal BCR transcriptional program specific for leukemic cells and the more aggressive cells.
Marker gene product functional analysis
Using the data generated from the gene expression profile over time, we identified genes with significant differences in expression over time and across the different cell populations after BCR stimulation. This identified a core BCR stimulation profile. We then superimposed significant differences between healthy and CLL B cells and then between indolent and aggressive CLL cells. The goal was to organize this gene expression data in biologically relevant pathways, and we used Pathways Ingenuity Analysis (Ingenuity Systems) software (http://www.ingenuity.com/products/pathways_analysis.html) to search for functionally related groups of genes across time and cell populations to identify interactions and further refined this gene product analysis using public databases and literature learning. Among the functional groups of genes identified, the most prominent centered on MYC and NFκB at later time points. Shown in Figure 4 are the changes in gene expression that occur after BCR stimulation in these functional pathways. Further validation of this approach comes from comparison of the genes identified in this analysis in human B cells with that previously reported in murine B cells.5 This analysis demonstrated that many of these core functionally related genes were identified in both analyses (Figure 4). At the earliest time point shown (60-90 minutes), genes involved in regulation of JUNB and FOS are up-regulated, with later up-regulation of MYC and NFκB1. Genes differentially regulated in CLL compared to healthy B cells in these pathways appear mainly to be involved in regulation of a balance between cell proliferation and apoptosis. For example, RAN, a Ras family member that is differentially expressed in CLL cells compared to healthy B cells, and YWHAQ, a 14-3-3 family member gene that is differentially expressed in aggressive compared to indolent CLL cells, participate in controlling cell-cycle progression. Several gene products are also involved in RNA processing, including NOL5A, EIF3S9, and FBL. Importantly, specifically in the CLL cells but not in the healthy B cells, there was up-regulation of other genes products that participate in regulating apoptotic function, such as PPARBP, RANBP2, and HNRPD, as well as proteasomal proteolysis including PSMA1 and PSMA2. These findings are in keeping with the functional data that there is a complex deregulation of numerous factors controlling balance between life and death within the leukemic cells compared to healthy B cells following BCR stimulation.

Graphical view of selected gene products based on Ingenuity Pathways Analysis. A cell is shown as an oval (the inner oval represents the nuclear membrane, the 2 outer ovals represent the plasma membrane) depicted at 4 different time points chosen for analysis (60 minutes, 90 minutes, 210 minutes, 390 minutes). Gene products in gray are located according to their respective known function within in the cell graph. Interaction edges between gene products are derived from Ingenuity Systems learning results. Expressed genes at each time point are represented successively in yellow (expressed in every healthy and malignant B cell), orange (differentially expressed between CLL and healthy B cells), and red (differentially expressed between aggressive CLL and indolent CLL). The expression of 3 main different functional group of genes (early transcriptional factors, MYC and NFκB) is represented in the graph across time. The malignant and aggressive malignant specific genes are integrated in these common functional groups of expressed genes and interfere mainly with the life and death balance at multiple levels.
Graphical view of selected gene products based on Ingenuity Pathways Analysis. A cell is shown as an oval (the inner oval represents the nuclear membrane, the 2 outer ovals represent the plasma membrane) depicted at 4 different time points chosen for analysis (60 minutes, 90 minutes, 210 minutes, 390 minutes). Gene products in gray are located according to their respective known function within in the cell graph. Interaction edges between gene products are derived from Ingenuity Systems learning results. Expressed genes at each time point are represented successively in yellow (expressed in every healthy and malignant B cell), orange (differentially expressed between CLL and healthy B cells), and red (differentially expressed between aggressive CLL and indolent CLL). The expression of 3 main different functional group of genes (early transcriptional factors, MYC and NFκB) is represented in the graph across time. The malignant and aggressive malignant specific genes are integrated in these common functional groups of expressed genes and interfere mainly with the life and death balance at multiple levels.
Quantitative gene expression and protein validation
We validated these results using Taqman RQ-PCR for selected genes in these functional pathways using an aliquot of the total RNA previously hybridized on the microarrays. Results from microarray and quantitative PCR for 3 representative genes (DUSP1, EGR2, NFκB1) with variable patterns of expression across subjects and across time are shown in Figure 5A and demonstrate an identical pattern across patients and over time, with correlation coefficients between microarray expression and RQ-PCR copy number between 0.8 and 0.87 and with variance higher at higher expression levels (Figure S1, available on the Blood website; see the Supplemental Figure link at the top of the online article). In addition, protein expression of NFκB1 and DUSP1 demonstrates differential expression with an increase in protein level of the p105 precursor and p50 NFκB1 and DUSP1 at different time points in the BCR stimulated compared to the unstimulated CLL cells, (Figure 5B) in keeping with the patterns of gene expression observed.

RNA and protein expression changes after BCR crosslinking. (A) Temporal expression of 3 representative genes (DUSP1, EGR2, NFκB1) in healthy B cells and CLL B cells assessed by gene expression profile and by RQ-PCR analysis. (B) Immunoblot analysis of NFκB1 and DUSP1 proteins in representative CLL sample. For immunoblot analysis, cell lysates were prepared from negatively isolated fresh primary B CLL cells (US or S) at 3 to 4 different time points after BCR stimulation (60, 210, and 390 minutes and 16 hours). Thirty micrograms of protein lysate from each sample was loaded onto a separate lane of SDS-polyacrylamide gel. The immunoblot was probed with anti-NFκB1 antibody to detect NFκB1 p50 protein and its p105 precursor protein (labeled arrows, left). Blots were reprobed with Tim 23 or GAPDH antibody as control (labeled arrow, bottom). The molecular mass of protein standards is shown.
RNA and protein expression changes after BCR crosslinking. (A) Temporal expression of 3 representative genes (DUSP1, EGR2, NFκB1) in healthy B cells and CLL B cells assessed by gene expression profile and by RQ-PCR analysis. (B) Immunoblot analysis of NFκB1 and DUSP1 proteins in representative CLL sample. For immunoblot analysis, cell lysates were prepared from negatively isolated fresh primary B CLL cells (US or S) at 3 to 4 different time points after BCR stimulation (60, 210, and 390 minutes and 16 hours). Thirty micrograms of protein lysate from each sample was loaded onto a separate lane of SDS-polyacrylamide gel. The immunoblot was probed with anti-NFκB1 antibody to detect NFκB1 p50 protein and its p105 precursor protein (labeled arrows, left). Blots were reprobed with Tim 23 or GAPDH antibody as control (labeled arrow, bottom). The molecular mass of protein standards is shown.
Balance between survival and death in leukemic cells
These data demonstrate how the more aggressive leukemic cells show a complex disorder of numerous genes regulating cell life and death. To test this hypothesis further, we measured the functional implications of these complex BCR transcriptional programs by monitoring survival and apoptosis in leukemic and healthy B cells after BCR stimulation (Figure 6). The basal rate of spontaneous apoptosis ranged from 6% to 16% (median, 9%) in healthy B cells and from 3% to 20% (median, 7%) in CLL cells. To appreciate the apoptotic response after BCR stimulation over 24 hours, we compared the number of cells undergoing higher Annexin V[e]FITC staining between S and control US cells for each patient at the same time point. The characteristics of the 12 cases examined are shown in Table 1. We used the modified histogram subtraction technique (Overton)29 to calculate the percentage of differentially stained cells before and after stimulation. At 6 hours after BCR engagement, the (S-US) Overton ranged from 19.6% to 33.6% (median, 26.8%) for the indolent CLL samples and 20.5% to 53.2% (median, 41.7%) for the aggressive CLL samples compared to from 1.8 to 29.6% (median, 14.4%) for the healthy donor controls. These finding are in keeping with the data in Figure 4 showing the up-regulation of genes involved in apoptosis in the more aggressive CLL samples. It is surprising that the more aggressive CLL cells undergo more apoptosis than the indolent leukemic cells or healthy B cells, but this may reflect the importance of the dynamic BCR genetic program in the aggressiveness of the leukemia and that cells that survive BCR-mediated apoptosis may then have increased proliferative potential.

BCR cross-linking and apoptosis. (A) Apoptosis assessed by Annexin V–FITC staining for 1 healthy control and 2 CLL patients (nonresponder and high responder). (Top and bottom left) Time before stimulation (T0). (upper right) Overlay unstimulated (T0) and stimulated (T6H, in translucent blue). (Bottom right) Integration of the 2 curves unstimulated and stimulated (T6H), calculation of the percentage of cells undergoing apoptosis after stimulation compared to basal (Overton). (B) Flow cytometry analysis of Annexin V–FITC and propidium iodine double staining before stimulation (T0), at 6 hours, and at 24 hours after BCR stimulation of a representative CLL high responder. Cells remains viable (Annexin V positive/PI negative) at 6 hours after stimulation.
BCR cross-linking and apoptosis. (A) Apoptosis assessed by Annexin V–FITC staining for 1 healthy control and 2 CLL patients (nonresponder and high responder). (Top and bottom left) Time before stimulation (T0). (upper right) Overlay unstimulated (T0) and stimulated (T6H, in translucent blue). (Bottom right) Integration of the 2 curves unstimulated and stimulated (T6H), calculation of the percentage of cells undergoing apoptosis after stimulation compared to basal (Overton). (B) Flow cytometry analysis of Annexin V–FITC and propidium iodine double staining before stimulation (T0), at 6 hours, and at 24 hours after BCR stimulation of a representative CLL high responder. Cells remains viable (Annexin V positive/PI negative) at 6 hours after stimulation.
Characteristics of CLL patients studied
| CLL patient label | Apoptotic response S-US at 6 h | IgVH status (%) | Zap-70 status | Cytogenetics |
|---|---|---|---|---|
| Indolent | ||||
| CLL-M4 | 21.2 | M-CLL VH3-23 (93) | Negative | Normal |
| CLL-M5 | 24.8 | M-CLL VH3-22 (87) | Negative | 13q− |
| CLL-M6 | 19.6 | M-CLL VH3-23 (92) | Negative | 13q− |
| CLL-M7 | 28.8 | M-CLL VH4-31 (86) | Negative | 13q− |
| CLL-M3 | 32.5 | M-CLL VH3-48 (91) | Negative | 13q− |
| CLL-M8 | 33.6 | M-CLL VH4-34 (94) | Negative | 13q− |
| CLL-M1 | ND | M-CLL VH4-31 (95) | Negative | 13q− |
| Aggressive | ||||
| CLL-UM1 | 20.5 | UM-CLL VH1-69 (99) | Positive | Add 12 |
| CLL-UM2 | 41.7 | UM-CLL VH1-69 (100) | Positive | t(8;13), add8, 11q− |
| CLL-UM3 | 41.7 | UM-CLL VH1-69 (100) | Positive | Add 12 |
| CLL-UM4 | 53.2 | UM-CLL VH4-34 (100) | Positive | Normal |
| CLL-M2 | 44.7 | M-CLL VH3-48 (97) | Positive | Normal |
| CLL patient label | Apoptotic response S-US at 6 h | IgVH status (%) | Zap-70 status | Cytogenetics |
|---|---|---|---|---|
| Indolent | ||||
| CLL-M4 | 21.2 | M-CLL VH3-23 (93) | Negative | Normal |
| CLL-M5 | 24.8 | M-CLL VH3-22 (87) | Negative | 13q− |
| CLL-M6 | 19.6 | M-CLL VH3-23 (92) | Negative | 13q− |
| CLL-M7 | 28.8 | M-CLL VH4-31 (86) | Negative | 13q− |
| CLL-M3 | 32.5 | M-CLL VH3-48 (91) | Negative | 13q− |
| CLL-M8 | 33.6 | M-CLL VH4-34 (94) | Negative | 13q− |
| CLL-M1 | ND | M-CLL VH4-31 (95) | Negative | 13q− |
| Aggressive | ||||
| CLL-UM1 | 20.5 | UM-CLL VH1-69 (99) | Positive | Add 12 |
| CLL-UM2 | 41.7 | UM-CLL VH1-69 (100) | Positive | t(8;13), add8, 11q− |
| CLL-UM3 | 41.7 | UM-CLL VH1-69 (100) | Positive | Add 12 |
| CLL-UM4 | 53.2 | UM-CLL VH4-34 (100) | Positive | Normal |
| CLL-M2 | 44.7 | M-CLL VH3-48 (97) | Positive | Normal |
Indolent: average, 26.8; median, 26.8. Aggressive: average, 40.4; median, 41.7.
Apoptotic response measured by flow cytometry after Annexin V–FITC and propidium iodide double staining, with percentage of BCR-stimulated cells compared to unstimulated cells undergoing Annexin V[e]FITC staining 6 hours after BCR cross-linking. Cytogenetics characteristics were assessed by FISH.
UM indicates IgVH unmutated; M, IgVH mutated; and ND, not determined.
Discussion
Here we used BCR cross-linking as a model to examine differential functional and transcriptional consequences in healthy and malignant B cells. We chose to examine this in CLL, where there is evidence to suggest an antigen-driven process and where the Ig mutational status has prognostic significance. We demonstrate that gene expression profiling over time not only identified transcription of genes, such as JUN and FOS, previously known to be involved in the regulation of BCR-mediated signaling but also identified novel genes and pathways involved in the early and late regulation of events. This analysis identified a core BCR gene expression pattern over time common to both healthy and malignant B cells, but it also identified differential regulation of BCR-mediated events in healthy compared to malignant B cells in genes involved mainly in regulating cell-cycle progression and apoptosis.
Any gene expression profiling performed over time must first identify the critical time points required for analysis. Our pilot study identified 4 time points that appeared most critical to capture changes in transcription over time. Even with only 4 time points examined and analysis of a relatively small number of samples, this required analysis of 144 arrays, and it is possible that we missed events important in regulating BCR-mediated signaling pathways by restricting analysis to these time points.
We developed a novel linear combination method for analysis to examine changes in gene expression over time. By creating a score of the weighted average of the BCR-stimulated effects over time, we were able to capture the overall BCR-signaling effect over time for each patient as a single score. To obtain gene marker lists discriminating 2 distinct groups defined by unsupervised clustering of this score, we adopted the method of Wei and Johnson30 and combined 4 individual tests for each time point. The dominant effect observed was the BCR signature, which was in keeping with our previous experience examining the consequences on global gene expression at a single time point after CD40 activation of healthy B cells and CLL cells.31 Within this present analysis, we could distinguish healthy from malignant B cells within the signatures of the stimulated cells, but even in this exploratory small set of patients we were further able to separate patients with aggressive CLL from those with indolent CLL. Of note, all the aggressive CLL samples expressed ZAP70, in keeping with studies demonstrating ZAP70 as an important regulator of BCR-mediated signaling in CLL.20,21
Usage of specific VH genes has a strong influence on gene expression, suggesting antigen recognition and ongoing BCR stimulation as important factors in the pathogenesis of CLL.32–34 On initial examination of the response to BCR cross-linking, it may seem counterintuitive that aggressive cases undergo more apoptosis, when in fact these cases have more aggressive clinical behavior. The fate of a cell after activation is dependent not only upon the receptor activated but also upon the strength of the signal delivered, a phenomenon that has been previously described for BCR stimulation in CLL that expresses CD3835 or ZAP70.20 Under physiologic conditions, where the CLL cells may encounter antigenic stimulation of the BCR, it is likely that coexpression of such modulatory molecules would also alter the fate of the cell, thereby contributing to the more aggressive clinical behavior, potentially by differential regulation of cell proliferation and apoptosis as shown in Figure 4. For example, after BCR stimulation, we identified increased expression of genes such as DUSP1 (MKP1), known to be involved in cell-cycle regulation36 and inhibition of which potentiates JNK-related apoptosis.37 Ongoing studies are examining the consequences of subsequent gene expression of down-regulation of DUSP1 using small interfering RNA after BCR activation. The number of samples analyzed for gene expression over time in this analysis is small, and the genetic heterogeneity of this disease and the interactions of VH mutational status and genetic background of the CLL cells32–34 is such that additional work will be necessary to differentiate the potential effects of BCR-mediated signaling events in all CLL cases. Although patient samples can be grouped on the basis of the functional consequence of BCR-mediated signaling, it is notable that even in those cases deemed not to be “competent” on BCR signaling, there are changes in gene expression over time, demonstrating that BCR-mediated signaling is possible in these cells, at least as measured after BCR crosslinking as performed here.
In conclusion, we demonstrate that it is possible to build temporal models of gene transcription using gene expression profiling and to use these to identify differences in signaling pathways in healthy compared to malignant cells using BCR cross-linking in healthy and malignant B cells as a model. Ongoing studies are examining the critical nodal points in genetic networks that regulate such events and the steps to be taken to modify the networks in malignant cells so that the functional consequences of signaling return to the physiologic rather than the pathophysiologic state.
Authorship
Contribution: J.G.G. designed and supervised the study. L.D.V. performed the experiments and analyzed data. Y.P. and C.L. performed statistical analysis. L.D.V. and J.G.G. wrote the manuscript. All authors have seen and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: John G. Gribben, Medical Oncology, Barts and The London School of Medicine, Charterhouse Square, London EC1M 6BQ, United Kingdom; e-mail: john.gribben@cancer.org.uk.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
This work was supported by the CLL Research Consortium PO1 CA 81538 from the National Cancer Institute (J.G.G.) and by the Eskandarian Family CLL Research Fund. L.D.V. was supported in part by Fondation de France (2003004300).
We thank Drs Edward A. Fox and Maura Berkeley (DFCI Microarray Core Facility, Boston, MA), Suzan Lazo (DFCI Flow Cytometry Core Facility, Boston, MA), and Laura Rassenti (CRC, La Jolla, CA) for determining the patients' ZAP70 status; Dr Frederic Davi, Pr Hélène Merle-Beral (Pitie-Salpetriere Hospital, Paris, France), and Dr Iozo Delic (CEA, Paris, France) for their support; Drs Emmanuel Zorn and Anne Astier (DFCI) for helpful advice; and Frederic Jacob for technical skill.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal