

In this issue of Blood, the mechanism of angioedema formation in hereditary angioedema (HAE) with mutant plasminogen is shown by Dickeson et al1 to be caused by the direct cleavage of kininogens by the mutant plasmin.
In 4 of the 6 types of HAE with normal C1 inhibitor (C1 INH), overproduction of bradykinin is the likely mediator of the angioedema. However, a detailed mechanism has been reported only for HAE with factor XII (FXII) mutation, the most common of the types worldwide (but rare in the United States). HAE with plasminogen mutation is the next most common.2,3 Dickeson et al demonstrate that a substitution of Lys311 for glutamic acid in plasminogen induces bradykinin generation by cleavage of kininogen once the plasminogen is converted to plasmin. Native, unmutated plasmin is significantly less effective in inducing generation of bradykinin. Although they did not address the in vivo activation of plasminogen directly, they added tissue plasminogen activator (tPA) to plasma and demonstrated enhanced bradykinin generation when plasminogen-Glu311 (Plm-Glu311) was present and converted to plasmin-Glu311 (Pln-Glu311). The result was strikingly similar upon tPA activation of plasma deficient in FXII or prekallikrein, to which Plm-Glu311 vs Plm-Lys311 was added, indicating that these proteins of the contact-activated bradykinin cascade are not necessary. The only other constituent of the latter cascade is kininogen, and the results point to a direct effect on kininogen. Because plasma has 2 kininogens, it was necessary to determine whether 1 or both are a substrate for Pln-Glu311.
High-molecular-weight kininogen (HK) is the kininogen specific to the plasma cascade and is cleaved by plasma kallikrein to generate bradykinin. It consists of a heavy chain at the N terminus, followed by bradykinin and then a unique light chain.4 Cleavage by plasma kallikrein at Arg-Ser (C-terminal site) and Lys-Arg (N-terminal site) liberates bradykinin, leaving heavy and light chains linked by disulfide bonds, known as kinin-free HK. The other form of kininogen, termed low-molecular-weight kininogen (LK) is not cleaved by plasma kallikrein. LK is digested by tissue kallikrein (secreted by cells) to liberate Lys-bradykinin, and plasma aminopeptidase P removes the N-terminal Lys. LK has the same heavy chain and bradykinin moiety as HK, but has a significantly smaller light chain. The light chains are added by splicing of alternative exons at a site 10 amino acids beyond the bradykinin moiety. The C-terminal domain of the light chain of HK binds either plasma prekallikrein or coagulation FXI, and this critical difference places HK in the plasma contact system,5 whereas LK is not part of the contact system. Dickeson et al tested the ability of Pln-Glu311 to cleave these kininogens. Both were activated to produce bradykinin (see figure). Since the concentration of LK exceeds that of HK in the plasma concentration by about three-fold, LK would be expected to be the major contributor to the formation of bradykinin, but it is likely that both have a role. The difference between LK and HK cleavage may reflect not only the direct effect upon each of them by Pln-Glu311, but whether in vivo plasmin production includes activation by the FXII–dependent fibrinolytic pathway. The latter possibility could involve classic contact activation of FXII or plasmin activation of FXII. The plasma kallikrein produced not only activates plasma prourokinase to convert plasminogen to plasmin (via the intrinsic fibrinolytic pathway), but also selectively cleaves HK to produce bradykinin.
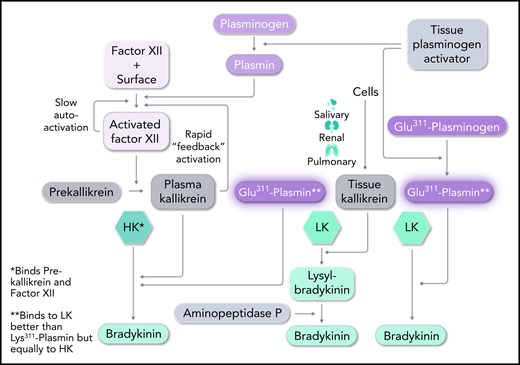
Pathways for bradykinin formation. In HAE, Plm-Glu311 conversion to Pln-Glu311 leads to direct digestion of both LK and HK to produce bradykinin. It essentially replaces tissue and plasma kallikreins in their action on LK and HK, respectively. C1 INH (mutated in HAE types 1 and 2) primarily inhibits FXIIa and plasma kallikrein, but weak inhibition of plasmin and tPA may have relevance to HAE-Plm, as well. Professional illustration by Somersault18:24.
Pathways for bradykinin formation. In HAE, Plm-Glu311 conversion to Pln-Glu311 leads to direct digestion of both LK and HK to produce bradykinin. It essentially replaces tissue and plasma kallikreins in their action on LK and HK, respectively. C1 INH (mutated in HAE types 1 and 2) primarily inhibits FXIIa and plasma kallikrein, but weak inhibition of plasmin and tPA may have relevance to HAE-Plm, as well. Professional illustration by Somersault18:24.
However, the superior response of patients to icatibant, a bradykinin B-2 receptor antagonist, compared with the response to C1 INH infusion (to supranormal levels)6 suggests a smaller contribution of the aforementioned plasma bradykinin-forming cascade. C1 INH is a weak inhibitor of tPA and plasmin. If tPA secretion were the initial source of plasmin formation, any effect of C1 INH could be separate from its inhibition of activated FXII or plasma kallikrein. The experiments with deficient plasma demonstrate that the latter 2 proteins are not requisite.
Finally, the other known form of HAE with normal C1 INH (HAE-N) has mutations of FXII, most commonly a replacement of Thr309 by Lys or Arg. The mutant protein lacks a carbohydrate attachment site and is more susceptible to plasmin digestion7 than is normal FXII.8 The product, a truncated FXII, is extremely sensitive to activation by plasma kallikrein,9 so the reciprocal activation of mutant FXII and prekallikrein is rapid (see figure). C1 INH no longer controls activation, and the resultant plasma kallikrein cleaves HK, to overproduce bradykinin.
As soon as a mutant plasminogen was shown to lead to HAE, the first assumption was that Plm-Glu311 may have an excessively rapid rate of activation to Pln-Glu311 and cleavage of native FXII, leading to excessive bradykinin formation. This is not the case. Dickeson et al showed that Pln-Glu311 and native plasmin cleave FXII at the same rate.10 Thus, the direct cleavage of kininogen(s) is primary.
Conflict-of-interest disclosure: The author declares no competing financial interests.


