In the current issue of Blood, describe the fate of FLT3-internal tandem duplication (ITD) clones in acute patients with myeloid leukemia (AML) treated with midostaurin.1
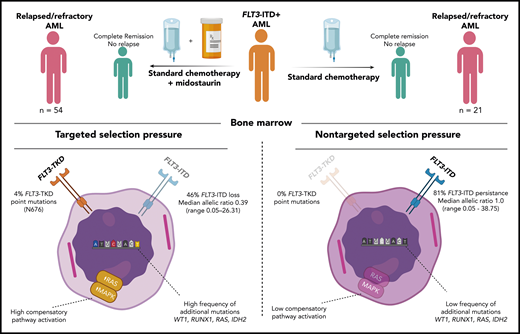
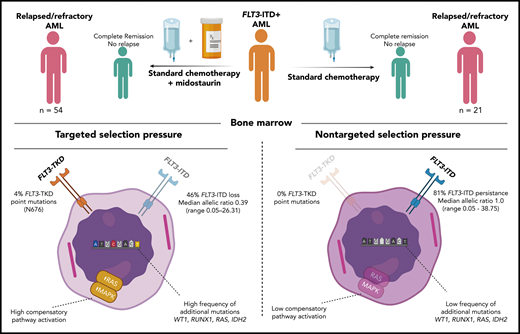
Molecular analyses of diagnosis and disease progression samples from 54 FLT3-ITD–positive, relapse/refractory patients treated with midostaurin addition (left) compared with those from 21 FLT3-ITD–positive, relapse/refractory patients not exposed to midostaurin (right). TKD, tyrosine kinase domain. Figure prepared by David G. J. Cucchi (created with BioRender.com).
Molecular analyses of diagnosis and disease progression samples from 54 FLT3-ITD–positive, relapse/refractory patients treated with midostaurin addition (left) compared with those from 21 FLT3-ITD–positive, relapse/refractory patients not exposed to midostaurin (right). TKD, tyrosine kinase domain. Figure prepared by David G. J. Cucchi (created with BioRender.com).
Current targeted treatment strategies have benefitted greatly from extensive genetic analyses to identify potential successful treatment targets, yet many targets are “moving,” and AML is a very heterogeneous disease harboring many clones with varying mutations.2 As for FLT3-ITD, the value of the target is clearly evident, as it is present in ∼25% of AML and confers a relatively poor prognosis.3 The RATIFY (Randomized AML Trial In FLT3 in patients less than 60 Years old) study did show that the addition of midostaurin (a first-generation nonspecific FLT3 mutation inhibitor) to standard chemotherapy offers a survival benefit. However, FLT3-ITD is often not a driver mutation, so leukemia cells may not depend on the mutation for their malignant behavior. Moreover, this target has been shown to be frequently unstable during disease progression.4 Achieving a complete response is currently not the major problem in AML but keeping patients in remission (ie, preventing relapse which still occurs in over 50% of patients) would improve outcome enormously. Understanding the mechanisms of relapse are in this respect of the utmost importance. Therefore, it is essential to understand the evolution of FLT3-ITD AML clones when patients receive treatment targeting this mutation. This study of Schmalbrock et al sheds light on the clonal selection under midostaurin treatment by comparing the evolution of FLT3-ITD clones in 54 relapsed/refractory patients who received the pan-kinase inhibitor midostaurin with a control group of 21 relapsed refractory patients who did not receive this drug during standard treatment (RATIFY and several German-Austrian Acute Myeloid Leukemia Study Group trials). Samples at diagnosis were compared with disease progression (refractory or relapse) samples for FLT3-ITD presence, allelic ratio and additional mutations as assessed by whole-exome sequencing. FLT3 inhibitor treatment resulted in more FLT3-ITD negative cases in the midostaurin treated patients (46%) compared with the control group (19%). Patients with persistent FLT3-ITD showed lower allelic ratios in the midostaurin group compared with control. Besides the two groups of FLT3-ITD loss and persistence, there were also patients who showed a change in ITD at relapse that was not seen in refractory patients, indicating the necessity of prolonged exposure for clonal selection (see figure). The targeted treatment obviously renders a selection pressure on these clones of which only the fittest survive.
Interestingly, in the study of Schmalbrock et al, only 2 patients developed a FLT3-resistant point mutation, which might be related to the fact that midostaurin is not specific for FLT3 but a pan-kinase inhibitor. This feature of midostaurin is potentially beneficial resulting in cell kill of FLT3-ITD–negative cells as well.5 In this respect, debate is ongoing about the benefits of multitargeted drugs like midostaurin or sorafenib compared with more specific targeted drugs like gilteritinib.6 Therefore, midostaurin and gilteritinib are being studied in a head-to-head comparison for both induction and maintenance in the HOVON 156/AML-SG 28-18 study (EudraCT #018000624-33).
The FLT3-ITD loss samples showed increase of RAS and MAPK pathway induction, which has been described previously as a cause for tyrosine kinase inhibitor resistance.7 The FLT3-ITD persistent samples revealed another category of genes related to cell cycle regulation. The fact that the FLT3 mutation was often still present at disease progression, despite the targeted treatment, suggests that some cells were inherent resistant and survived. However, it cannot be ruled out that drug dosing, compliance, and distribution-related mechanisms also played a role. Plasma inhibitory activity, which was measured in a subset of patients (n = 12), was highly variable, and several patients had to stop treatment due to side effects, which may have let to insufficient dosing. Other limitations of this study are the relatively insensitive (5%) FLT3-ITD measurement, by which small FLT3-ITD subclones may have been missed, and the uncertainty of the influence of allogenic stem cell transplantation on the mutational profile of the clones due to low numbers of patients. Many other factors that could be of importance for the development or selection of mutation, such as conditioning regimen before transplant, source of stem cells, graft-versus-host prophylaxis, rapidity of achieving remission, and use of maintenance treatment, have to be investigated in future studies.
Yet, this study provided important insights in the clonal architecture of AML after midostaurin treatment. Although the control group is small, it shows in a comparable cohort that without midostaurin the patients have a greater persistence of FLT3-ITD clones with a relatively higher allelic ratio. Monitoring recurrent AML mutations in addition to the FLT3-ITD, during therapy has been shown to be essential. Large add-on studies to clinical trials, such as those described here, are a substantial effort but provide critical data to optimize and personalize the appropriate targeted agents in this oligoclonal disease. The current data imply a constant war on the upcoming clones that are selected by “survival of the fittest.” Instruments of war against AML include changing of the treatment according to the upcoming dominant clones (eg, WT1, RUNX1, RAS, and IDH2). In particular, in complete remission, the more sensitive possibility of measurable residual disease detection (by flow cytometry and molecular techniques) may offer the possibility of personalized targeted early intervention.8 Another approach that can also be deduced from these results is upfront combination therapy for cells that can use alternative proliferative pathways besides the FLT3 pathway.9 Ongoing studies that add FLT3 inhibitors to low-intensity treatment may also change the mutational landscape. In the near future, further insights into clonal architecture will come from single-cell analyses showing possible biomarkers of clonal profiles that may identify patients eligible for a specific approach.10 Schmalbrock and colleagues paved the way for such a promising future.
Conflict-of-interest disclosure: The authors declare no competing financial interests.