

In this issue of Blood, expand our understanding of thrombosis and bleeding risks and their predictors in hospitalized patients with coronavirus disease 2019 (COVID-19).1
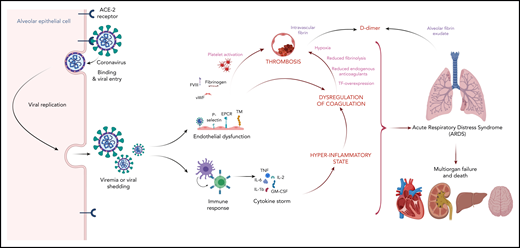
Potential mechanism linking dysregulated inflammation and coagulation with thrombosis. SARS-CoV-2 infects respiratory epithelial cells by binding to the angiotensin-converting enzyme 2 (ACE-2) receptor. Shed virus elicits an inflammatory response, which can be maladaptive in some cases leading to a cytokine storm mediated by proinflammatory cytokines such as interleukin 1β (IL-1β), IL-2, IL-6, tumor necrosis factor (TNF), and granulocyte-macrophage colony-stimulating factor (GM-CSF). Proinflammatory cytokines and SARS-CoV-2 infection of the endothelium contribute to hypercoagulability by upregulation of procoagulants such as tissue factor (TF), P-selectin, factor VIII (FVIII), fibrinogen, and von Willebrand factor (vWF); downregulation of anticoagulants such as thrombomodulin (TM) and endothelial protein C receptor (EPCR); and modulation of fibrinolysis by increased expression of type 1 plasminogen activator inhibitor, and leukocyte recruitment. Finally, the hypoxia and immobility in hospitalized patients with COVID-19 are potent triggers of thrombosis. Illustration created with BioRender.com.
Potential mechanism linking dysregulated inflammation and coagulation with thrombosis. SARS-CoV-2 infects respiratory epithelial cells by binding to the angiotensin-converting enzyme 2 (ACE-2) receptor. Shed virus elicits an inflammatory response, which can be maladaptive in some cases leading to a cytokine storm mediated by proinflammatory cytokines such as interleukin 1β (IL-1β), IL-2, IL-6, tumor necrosis factor (TNF), and granulocyte-macrophage colony-stimulating factor (GM-CSF). Proinflammatory cytokines and SARS-CoV-2 infection of the endothelium contribute to hypercoagulability by upregulation of procoagulants such as tissue factor (TF), P-selectin, factor VIII (FVIII), fibrinogen, and von Willebrand factor (vWF); downregulation of anticoagulants such as thrombomodulin (TM) and endothelial protein C receptor (EPCR); and modulation of fibrinolysis by increased expression of type 1 plasminogen activator inhibitor, and leukocyte recruitment. Finally, the hypoxia and immobility in hospitalized patients with COVID-19 are potent triggers of thrombosis. Illustration created with BioRender.com.
COVID-19, which is caused by severe acute respiratory syndrome coronarvirus 2 (SARS-CoV-2), has spread across the globe. Although most patients recover within 1 to 3 weeks, COVID-19 has already caused >100 000 deaths in the United States alone. SARS-CoV-2 enters cells by binding to the angiotensin-converting enzyme 2 receptor, which is expressed on respiratory epithelial cells and other cell types, including endothelial cells. Unchecked viral replication induces a florid host response characterized by dysregulation of inflammation and coagulation. Dysregulation of coagulation produces a coagulopathy associated with hypercoagulability as evidenced by venous and arterial thrombosis and multiorgan dysfunction (see figure). Up to 20% of affected patients require hospitalization, and the mortality rate in such patients is high.
The coagulopathy associated with COVID-19 is characterized by mild thrombocytopenia, slight prolongation of the prothrombin time, high levels of D-dimer, and elevated levels of fibrinogen, factor VIII, and von Willebrand factor. The levels of D-dimer, a breakdown product of cross-linked fibrin, correlate with disease severity and predict the risk of thrombosis, the need for ventilatory support, and mortality.
Considerable evidence indicates that COVID-19 is associated with a hypercoagulable state. Thus, despite anticoagulant thromboprophylaxis, studies from The Netherlands, France, and Italy have reported rates of venous thromboembolism (VTE) and arterial thrombosis ranging from 15% to 30% in critically ill patients with COVID-19 and ∼7% in those admitted to medical wards.2-4 Clotting of access catheters, dialysis membranes, and extracorporeal circuits has also been reported. Furthermore, in patients dying from COVID-19, autopsy studies reveal unsuspected deep vein thrombosis and multiple thrombi in the vessels of the lungs, kidneys, and other organs.5 These findings have prompted some clinicians to use treatment doses of heparin or low-molecular-weight heparin instead of prophylactic doses in critically ill COVID-19 patients.
Despite the wealth of clinical information, there are problems with the studies available to date. These include their mainly retrospective design, short follow-up, and diagnostic bias because some studies screened for VTE or included suspected but unconfirmed events, whereas others did not. Furthermore, interpretation is complicated because (a) the criteria for hospitalization of patients with COVID-19 and for admission to intensive care units vary, (b) treatments have evolved over time, and (c) differences in comorbidities and care settings may influence the rates of thrombosis. Therefore, more data are needed.
To add to our knowledge base, Al-Samkari et al report the results of a multicenter retrospective study that included 400 hospitalized patients diagnosed with COVID-19 between 1 March and 5 April 2020 at 5 Partners Healthcare institutions in Massachusetts. Rates of thrombosis, bleeding, and mortality were captured, and the prognostic value of markers of inflammation (C-reactive protein, erythrocyte sedimentation rate, ferritin, and procalcitonin) and coagulation (D-dimer, fibrinogen, prothrombin time, activated partial thromboplastin time, and platelet count) measured at presentation was examined.
Although some findings are confirmatory, the authors provide observations that challenge previous data or provide new information. Consistent with prior reports, this study confirms that COVID-19 is associated with an inflammatory state and a coagulopathy that are associated with high rates of venous and arterial thrombosis, progression to critical illness, and mortality. Of the 400 hospitalized patients, 144 patients (36%) became critically ill, 38 patients (9.5%) had confirmed or suspected venous or arterial thrombotic events, and 19 patients (4.8%) had documented VTE despite routine anticoagulant thromboprophylaxis. Although many of the markers of inflammation and coagulation correlated with clinical outcomes, D-dimer emerged as the most useful. Thus, D-dimer levels over 2500 ng/mL, levels over 5 times higher than the upper limit of normal, were associated with sevenfold, twofold, and 15-fold increases in the risk of thrombosis, progression to critical illness, and mortality, respectively.
What are the new findings? First, the 7.6% rate of VTE in critically ill COVID-19 patients in this study is lower than what has previously been reported and is more in line with the rate of VTE found in critically ill patients without COVID-19.6 Second, with anticoagulant thromboprophylaxis, the authors report a rate of major bleeding of 5.6% in critically ill patients with COVID-19 and identify a baseline platelet count below 150 × 109/L and D-dimer levels over 2500 ng/mL as independent predictors of a threefold increase in the risk of major bleeding.
The strengths of this study include the relatively large sample size and the comprehensive reporting of clinical outcomes. However, like many previous studies in patients with COVID-19, there are limitations, including the retrospective design as well as the potential for bias in case ascertainment and underestimation of VTE rates because of the inability to image all critically ill patients with suspected events.
The triggers responsible for COVID-19–associated coagulopathy remain elusive. Potential triggers include cytokine-induced overexpression of tissue factor, endothelial dysfunction with loss of its antithrombotic phenotype, stasis, and hypoxia (see figure). This study confirms the correlation between markers of inflammation and coagulation and supports the concept that inflammation is a major driver of the hypercoagulable state. An inflammation-driven hypercoagulable state has also been reported in critically ill patients with viral pneumonia caused by H1N1 or SARS-CoV-1.7 The VTE rate in such patients ranged from 5% to 25%, which is similar to the rates observed in patients with COVID-19.8-10 Although intensified anticoagulation regimens may reduce the risk of a thrombotic event, the results of this study raise the possibility that they may increase major bleeding rates to unacceptable levels in critically ill patients. As the world waits for the second wave of COVID-19, randomized trials comparing anticoagulation dosing strategies are urgently needed. Fortunately, several such trials are under way.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

