In this issue of Blood, contribute to overhauling the molecular classification system for diffuse large B-cell lymphoma (DLBCL).1 Two decades ago, gene expression profiling (GEP) enabled the division of DLBCL into 2 cell-of-origin (COO) molecular subgroups, a stratification with prognostic implications (see figure).2 This stimulated the search for therapies that could exploit the unique molecular features underlying each subgroup, long before the concept of “precision medicine” had gained popularity. The eventual failure of numerous clinical trials of targeted therapies selecting patients using COO implies that this classification, although foundational, lacks sufficient granularity to serve this purpose.

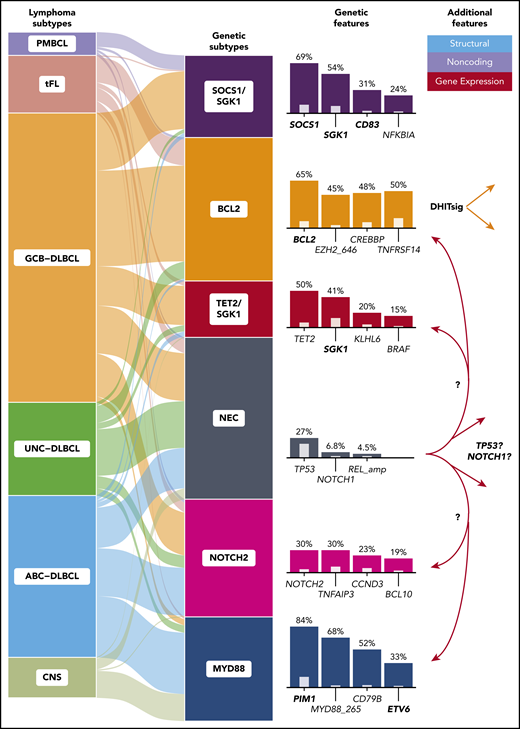
The DLBCL subtypes and molecular subgroups (left) form clusters with imbalanced representation of COO and other aggressive B-cell lymphomas (middle). Genes most frequently mutated in each cluster are indicated (right, colored bars) along with their relative enrichment when compared with the other groups (inner bars). For genes affected by aSHM (boldface), the presence of any coding or noncoding mutation was used for clustering. Three of these clusters closely resemble those identified previously: the germinal center B-cell (GCB)-enriched BCL2 cluster (or EZB/C3) and the activated B-cell (ABC)-predominant MYD88 cluster (MCD/C5), and the NOTCH2 cluster, which was also previously identified (BN2/C1) and comprises a balance of COO subgroups. The NEC cluster represents tumors lacking sufficient features to allow inclusion elsewhere. The TET2/SGK1 and SOCS1/SGK1 clusters were both newly identified and are both enriched for SGK1 mutations. DHITsig indicates GCB-DLBCL either positive or negative for the double-hit gene expression signature.10 CNS, central nervous system; tFL, transformed follicular lymphoma; UNC, unclassifiable.
The DLBCL subtypes and molecular subgroups (left) form clusters with imbalanced representation of COO and other aggressive B-cell lymphomas (middle). Genes most frequently mutated in each cluster are indicated (right, colored bars) along with their relative enrichment when compared with the other groups (inner bars). For genes affected by aSHM (boldface), the presence of any coding or noncoding mutation was used for clustering. Three of these clusters closely resemble those identified previously: the germinal center B-cell (GCB)-enriched BCL2 cluster (or EZB/C3) and the activated B-cell (ABC)-predominant MYD88 cluster (MCD/C5), and the NOTCH2 cluster, which was also previously identified (BN2/C1) and comprises a balance of COO subgroups. The NEC cluster represents tumors lacking sufficient features to allow inclusion elsewhere. The TET2/SGK1 and SOCS1/SGK1 clusters were both newly identified and are both enriched for SGK1 mutations. DHITsig indicates GCB-DLBCL either positive or negative for the double-hit gene expression signature.10 CNS, central nervous system; tFL, transformed follicular lymphoma; UNC, unclassifiable.
Over the last decade, large sequencing efforts have yielded a compendium of genetic aberrations common in DLBCL, with many of these exhibiting segregation by COO.3,4 Although this lends credence to molecular differences underlying the subgroups, the heterogeneity implied a hierarchy of genetically distinct entities. In 2018, two studies proposed a completely new framework for DLBCL subclassification relying on tumor genetics.5,6 They each suggested that DLBCL can be divided into at least 4 genetic clusters with some of these strongly enriched for cases representing either COO subgroup (see figure). Although broadly consistent in their conclusions, there were also significant discrepancies, leaving the nature of the DLBCL genetic subgroups unresolved.
The study by Lacy et al affirms the biological validity of 3 of the previously postulated clusters (see figure) and highlights methodological differences that likely underlie discrepancies between genetic classifications. They applied a distinct clustering method to mutation and sparse copy number data from 928 patients, mainly representing cases of de novo DLBCL and identified 6 clusters, which they named according to the most prevalent genetic feature(s) (see figure). As had been shown previously, primary central nervous system lymphomas were mostly assigned to a cluster typified by MYD88 mutations. Based on genetic features shared with marginal zone lymphoma, the second cluster may represent cases of occult transformation. Similarly, the third cluster contains many of the transformed follicular lymphoma cases and is enriched for BCL2 and EZH2 mutations. Importantly, one of the novel clusters (“not elsewhere classified," or NEC) acts as a sink for otherwise unclassifiable cases. This appears to mitigate the potential for misclassification of cases having limited defining features.
Although encouraging, these new results highlight the lack of equivalents for 2 clusters proposed in the aforementioned studies. This includes the largest cluster described by Chapuy et al, which was typified by TP53 aberrations and aneuploidies.5 Acknowledging the limitations of their feature set, Lacy et al applied their algorithm to the features used in the former study and confirmed that a comparable cluster could be identified by their method, given the availability of sufficient copy number features. Similarly, the group of tumors characterized by NOTCH1 mutations described by Schmitz et al has not yet been reproduced.6 The overall incidence of these mutations was low, and they were mildly overrepresented in NEC, possibly implying that this cluster could be identified by a more sensitive approach. Nonetheless, whether TP53-mutant tumors with aneuploidy and NOTCH1-mutant tumors share sufficient and unifying biology currently remains unresolved.
All genetic classifications proposed thus far rely heavily on mutations introduced by aberrant somatic hypermutation (aSHM). These are commonly noncoding and affect active promoters, 5′-untranslated regions (UTRs), introns, and enhancers as a function of transcriptional activity.4 Two of the new clusters are overrepresented for mutations in SGK1 and either TET2 or SOCS1, respectively (see figure). Although SGK1-mutant clusters were previously proposed, the further granularity provided by TET2 mutations is striking. Notably, this gene was recently identified as a tumor suppressor in DLBCL and was not used as a feature for the earlier genetic classifiers.7 The second SGK1-mutated cluster represents most of the primary mediastinal B-cell lymphomas (PMBCLs) along with DLBCLs without mediastinal involvement, which may highlight tumors that share biology with PMBCL. Considering the incomplete coverage of aSHM regions in the panel used by Lacy et al and the recent identification of new driver mutations, including a hot spot in the 3′-UTR of NFKBIZ, we speculate that additional noncoding mutations may further inform on genetic classification.
As we have established, varied use of features can lead to incomplete resolution of meaningful clusters. The approach used by Lacy et al overlooks the importance of structural variants through reliance on an exon panel, thus implicitly excluding important drivers such as translocations involving oncogenes. This class of drivers is so central to biology and prognosis that the World Health Organization has recognized high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements (HGBL-DH/TH) as a distinct entity.8 We have shown that GEP can recapitulate the biology of HGBL-DH/TH and provide more granular subdivisions within DLBCL that are not adequately explained by mutational data alone.9 We anticipate that future classifications could benefit from the explicit inclusion of structural variants and gene expression features to overcome some of the limitations of previous methods and enable further patient stratification.
Genetic classification represents a major step toward improved understanding of the biology of the heterogeneous entity we call DLBCL. The inclusion of features not queried by exon-centric sequencing panels has the potential to uncover further groups, refine existing groups, and minimize unclassified tumors. We predict that the saturation of informative features and genetic clusters will require additional comprehensive sequencing of a large number of tumors with a combination of whole-genome and transcriptome sequencing. This should allow reconciliation of the remaining disparities and lead to a unified framework on which we can begin to explore alternative treatments. Once the identities and characteristics of DLBCL genetic subgroups have been resolved, the community will ultimately require robust algorithms for sample classification. This will likely entail identification of a minimal set of features that can be identified within pathology laboratories in a timeframe suitable for clinical trials and, eventually, routine patient management. Over the past decade, attempts at improving patient outcomes beyond that achieved using standard immunochemotherapy using precision medicine have been disappointing. The next step in this exciting process is to demonstrate that this new molecular taxonomy can be translated into clinical benefit.
Conflict-of-interest disclosure: R.D.M. consults for Celgene and was named inventor on patents relating to DLBCL classification. D.W.S. consults for Abbvie, Celgene, and Janssen; receives research funding from Janssen, NanoString Technologies, and Roche/Genentech; and was named inventor on patents relating to DLBCL classification.