

Background: Recombination-activating gene (RAG) mediates recombination of the immunoglobulin heavy chain variable gene (IGHV) in immature B lymphocytes. Aberrant targeting of RAG to non-IGH sites in B cell acute lymphoblastic leukemia (B-ALL) contributes to the development of driver mutations and clonal evolution (Papaemmanuil et al. Nat. Genet. 2014). This finding suggests that patients whose ALL involves increased RAG-mediated clonal diversification would exhibit more aggressive disease. As an initial step toward addressing this hypothesis, we asked whether the extent of RAG-mediated diversification in a patient's leukemia is consistent across all cells or variable between subclones. To assess variation in RAG-mediated subclone diversification, we interrogated rearranged IGH sequences from diagnostic B-ALL specimens to identify early subclones (SC) and to quantify RAG-derived sub-subclones (SSC) from each. We hypothesized that if RAG activity is a consistent feature of the leukemia, all SC within a single patient will have a comparable extent of SSC evolution.
Methods: Amplicon-based IGHV sequencing identified the number of clonal IGH SC and their RAG-derived SSC in 22 pre-treatment adult and pediatric patients with newly-diagnosed B-ALL. Analysis was performed on peripheral blood (PB) for all patients in addition to diagnostic bone marrow (BM) for 16 of the 22 patients studied. Ultra-deep sequencing of IGH utilized 500 ng genomic DNA (representing approximately 80,000 cells/specimen) for the MiSeqDx platform, generating ~300 bp reads surrounding the VDJ junctional region. The NCBI IgBlast sequence analysis tool assigned IGH VDJ identities according to germline reference. Methods for determining subclones (SC) of shared clonal lineage involved classifying reads with a common Jh identity and 6 shared upstream Dh-Jh junctional nucleotides (termed "6N_Jx") according to defined methods (Gawad et al. Blood 2012). Further evolved sub-subclones (SCC) - as evidence of ongoing RAG activity - were defined by unique junctional sequences upstream of the common Dh-Jh junction (termed the "NDN" region). SSC were quantified according to their relative proportions within each SC family.
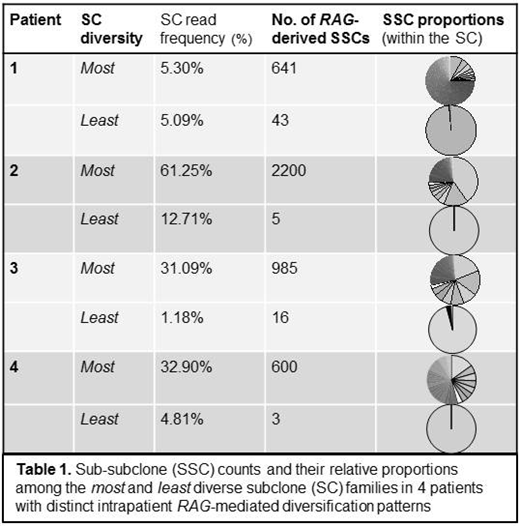
Results: VDJ-rearranged SC families were detected for 20 of the 22 patients studied (median 2/patient; range 1-7); further analysis to assess variation in RAG-mediated diversification was limited to the 18 cases with ≥ 2 SC. In 9 of these 18, numerous evolved SSCs were identified from at least one SC in the specimen. In 4/9, there were starkly distinct levels of RAG-mediated diversity observed between intrapatient SC families, with some clonal precursors giving rise to numerous SSCs (up to 2,200 SSC per SC) while others showed minimal-to-no RAG-derived evolution (Table 1). Fifteen of 16 patients with matched BM and PB diagnostic specimens had detectable SCs. Of these, 73% (11/15) shared the dominant SC, while in 27% (4/15) the dominant SC differed between sites. However, regardless of which SC predominated, the extent of SSC diversification within each SC was preserved between the BM and the PB, with similar evolution patterns observed regardless of disease site. There was no relationship between SC read frequency and number of SSCs.
Conclusions: Using deep sequencing of a single IGHV locus, wide variation in the extent of subclone diversification was observed in 4 of 20 patients with B-ALL. These findings indicate that the degree of RAG-mediated heterogeneity in B-ALL can range from minimal to extensive among distinct subclones in a single patient. The data underscore the relevance of single cell investigation of tumor characteristics to improving our understanding of the mechanisms of clonal evolution in lymphoid malignancies.
No relevant conflicts of interest to declare.

