

The Congenital Dyserythropoietic Anemia Registry (CDAR, ClinicalTrials.gov Identifier: NCT02964494) was created to investigate the natural history, biology, and molecular pathogenetic mechanisms of CDA. To date, there are 6 genes known to cause CDA (CDAN1, C15orf41, SEC23B, KIF23, KLF1, GATA1). However, 57% of patients registered in CDAR so far (17 out of 33 patients) have an unidentified genetic cause. We have utilized whole exome sequencing (WES) in family-trio design to search for novel candidate gene mutations that may be responsible for the disease. Three unrelated patients with dyserythropoiesis, hemolytic anemia, and neurodevelopmental delay were found to have missense mutations in the gene VPS4A which encodes an ATPase that participates with the ESCRT III machinery in endosomal vesicle trafficking, centrosome localization, and the abscission step of cytokinesis. It has been shown to play an essential role in division of HeLa cells in vitro where it concentrates at the spindle poles during mitosis and at the midbody during cytokinesis. The aim of this work is to validate the pathogenetic role of these VPS4A variants in CDA and further investigate the role of VPS4A in erythropoiesis.
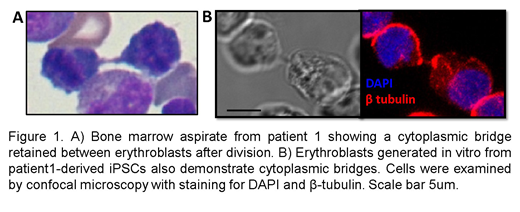
Patients 1 and 3 had de novo mutations (R284W and G203A) and transfusion-dependent anemia with presence of binucleated erythroblasts in the bone marrow resembling CDA type I. Of note, the patients' erythroblasts exhibited cytoplasmic bridges (Figure 1A) rather than the nuclear chromatin bridges observed in CDA-I. Patient 2, offspring of consanguineous parents, presented with hemolytic anemia and was found to have a homozygous mutation (A28V) in a highly conserved alanine residue in the microtubule-interacting domain (MIT) of VPS4A. She had rare evidence of dyserythropoiesis with fewer than 3% binucleated erythroblasts in bone marrow studies. All three patients had significant neurodevelopmental delay with axial hypotonia and appendicular hypertonia. Flow cytometry analysis of peripheral blood from each of these patients revealed a unique cell population which is negative for RNA (by thiazole orange) but still CD71 positive suggesting that loss of VPS4A function also impacts reticulocyte maturation, likely because of defective endosomal vesicle trafficking.
Using CD34+ cells in ex vivo erythropoiesis cultures, we first confirmed that VPS4A is expressed in human erythroblasts and localizes at the spindle poles and midbody during mitosis and cytokinesis in these cells. RNA isolated from reticulocytes from patients 1 and 2 was assessed for expression of VPS4A and the paralogous VPS4B. Samples from patient 1 had reduced expression of VPS4A (<50%) and only slight increase (2-3%) in VPS4B while samples from patient 2 had normal expression of VPS4A, but 10-20x increase in VPS4B highlighting the variable effects of these mutations on protein expression, function, and disease phenotype. Using iPSCs derived from patient 1 peripheral blood mononuclear cells (PBMCs), we have modeled the disease with in vitro erythropoiesis cultures. We observed decreased VPS4A expression and mislocalization in dividing erythroblasts produced from the patient-derived iPSCs. Moreover, the erythroblasts cultured from patient 1 demonstrated an increased rate of being binucleated and frequently maintained cytoplasmic bridges as seen in the patient's bone marrow (Figure 1B).
In summary, VPS4A plays a critical role in human erythroblast mitosis, cytokinesis, and reticulocyte maturation and mutations in VPS4A cause congenital dyserythropoietic and hemolytic anemia. The CDA registry is critical to allow systematic gene and pathway evaluation, enable novel treatment considerations, and reveal molecular mechanisms essential for erythropoiesis.
Kalfa:Agios: Other: local PI of clinical research trial; FORMA: Other: sponsored research agreement.

