

Juvenile myelomonocytic leukemia (JMML) is an aggressive, rare form of early childhood leukemia driven by Ras pathway mutations. Mutations in SET binding protein 1 (SETBP1) are a strong predictor of relapse in JMML, and are associated with reduced five-year event-free survival. Although some mechanisms of oncogenesis have been established for SETBP1 mutations, it remains unclear why they are associated with poor prognosis and relapse. The goal of this study was to understand how SETBP1 modulates the biology of Ras-driven leukemias and to determine whether there are therapeutic vulnerabilities of SETBP1-JMML that can be exploited. Here, we present novel findings on the synergy of SETBP1 and NRAS, and provide pre-clinical evidence for therapeutic intervention.
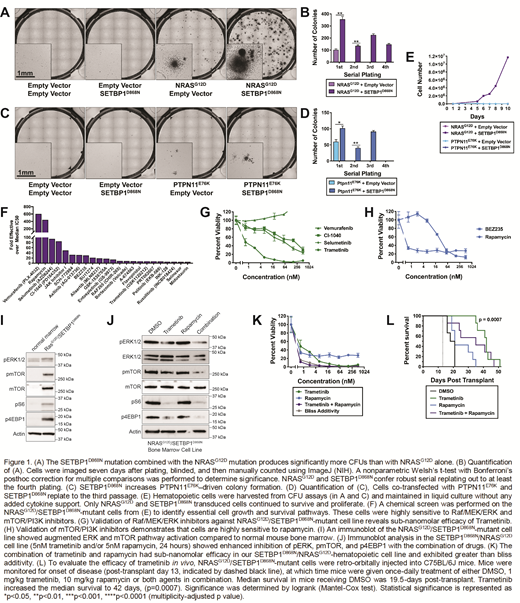
To address our central question of how SETBP1 mutations modulate Ras pathway-driven leukemia, we first set out to determine whether mutant SETBP1 promotes the growth of hematopoietic progenitors in the context of a Ras pathway mutation. To this end, we performed mouse hematopoietic colony forming unit assays in the absence of exogenous cytokines. Both NRAS-G12D and PTPN11-E76K alone formed a modest number of colonies, and the addition of SETBP1-D868N significantly augmented colony number with either Ras pathway mutation. The combination of NRAS-G12D and SETBP1-D868Nconfer robust serial replating, indicating that the SETBP1-D868N promotes oncogenic transformation and progenitor self-renewal in the NRAS-G12D mutant cells.
To understand how SETBP1 modulates therapeutic response, a novel NRAS/SETBP1-mutant cell line was generated and analyzed with a chemical screen of essential cell growth and survival pathways. This screen revealed dependencies on the mTOR/AKT/PI3K and Raf/MEK/ERK pathways. An immunoblot analysis revealed that mutant SETBP1 enhanced NRAS-driven ERK and mTOR pathway activation. Inhibitors of these pathways, such as rapamycin and trametinib were highly efficacious against our cell line. The combination of trametinib and rapamycin had sub-nanomolar efficacy in our NRAS/SETBP1-hematopoietic cell line and exhibited greater than bliss additivity at several time points.
To evaluate the efficacy in vivo, our SETBP1/NRAS-mutant cell line was injected into mice. At the onset of disease, mice were given once-daily treatment of trametinib, rapamycin, combination treatment, or DMSO control. The median survival of mice receiving DMSO was 19.5-days post-transplant, compared to 21 days for rapamycin, 35 days for the combination treatment, and 42 days for trametinib. Treatment with trametinib significantly increased the median survival to beyond rapamycin or DMSO, doubling the survival time of the mice.
Our data demonstrates that SETBP1 mutations accelerate NRAS-driven oncogenesis and enhance MAPK pathway activation by NRAS-G12D. Despite enhanced transforming potential, SETBP1-mutant cells are still sensitive to inhibitors of the RAS/ERK/MAPK pathway. Trametinib, an inhibitor of this pathway, doubles overall survival in our murine model of NRAS/SETBP1-mutant leukemia, thus providing encouraging pre-clinical data for the use of trametinib in SETBP1-mutant disease.
No relevant conflicts of interest to declare.

