In this issue of Blood, describe the in vitro and in vivo efficacy of a novel small molecule that induces the selective degradation of Bruton tyrosine kinase (BTK), IKZF1, and IKZF3, 3 proteins critical for the survival of many B-cell malignancies.1
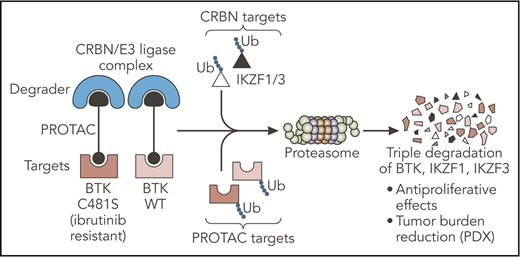
The PROTAC designed by Dobrovolsky et al leads to the triple degradation of BTK, IKZF1, and IKZF3 through their Ub and consequent degradation by the proteasome. This small molecule degrades both WT and C481S-mutated BTK proteins and overcomes ibrutinib resistance in B-cell malignancies in vivo. CRBN, cereblon; PDX, patient-derived xenograft; Ub, ubiquitination; WT, wild-type. Professional illustration by Somersault18:24.
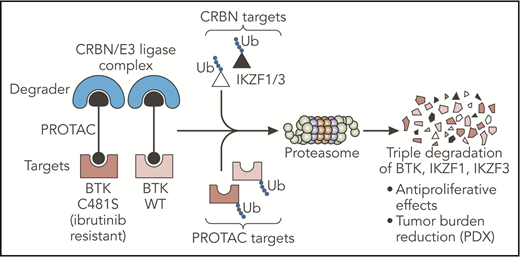
The PROTAC designed by Dobrovolsky et al leads to the triple degradation of BTK, IKZF1, and IKZF3 through their Ub and consequent degradation by the proteasome. This small molecule degrades both WT and C481S-mutated BTK proteins and overcomes ibrutinib resistance in B-cell malignancies in vivo. CRBN, cereblon; PDX, patient-derived xenograft; Ub, ubiquitination; WT, wild-type. Professional illustration by Somersault18:24.
The development of targeted therapies has recently opened a new area in the treatment of cancer. In aggressive B-cell malignancies, such as mantle cell lymphoma (MCL), selective inhibitors against BCL2 (venetoclax) or BTK (ibrutinib) gave unprecedented efficacy as single agents.2,3 Nevertheless, several mechanisms of intrinsic or adaptive resistance to these selective inhibitors, including activation of compensatory pathways and acquisition of mutations, have been recently described. In addition, retrospective studies revealed poor outcomes for ibrutinib relapsed/refractory MCL patients,4 highlighting an urgent need for novel therapeutic strategies preventing such drug resistance development.
Dobrovolsky et al designed a novel small molecule (DD-03-171) selective for the BTK protein, based on the proteolysis targeting chimeras (PROTACs) platform. PROTACs are composed of 2 arms, one used as a ligand for a specific target and the other as a ligand for a “degrader,” both tied up by a linker. This structure successfully targets specific proteins for ubiquitination and degradation by the proteasome. In the present study, the authors used a noncovalent inhibitor to target BTK and linked it with an immunomodulatory imide drug (IMiD), the well-described ligand for the cereblon/E3 ubiquitin ligase “degrader” complex. In contrast to inhibitors of kinase activity, this PROTAC led to the degradation of the BTK kinase and consequently targeted additional noncatalytic function of this protein. Importantly, binding of these small molecule degraders may occur noncovalently at any sites on the target. This property allowed the authors to design a molecule efficient for wild-type BTK but also for C481S BTK, a mutation described as impairing ibrutinib binding and efficacy and inducing subsequent drug resistance.5 In addition, the IMiD moiety present on the PROTAC led to the cereblon-dependent degradation of IKZF1/3, 2 essential transcription factors in B-cell malignancies.6 Taken together, Dobrovolsky et al designed a selective triple-degrader potentially able to overcome ibrutinib resistance (see figure).
Through a series of elegant experiments, the authors first demonstrated the molecular efficacy of DD-03-171 in several lymphoma cell lines and showed an antiproliferative effect, irrespective of cell sensitivity to ibrutinib. Given that ibrutinib resistance is a major issue in B-cell malignancies, the results shown in the present study are clinically relevant and of great interest for the field. This interest can also be measured by the publication of several recent reports evaluating the same PROTAC platform to target BTK in similar cellular models.7-9 Notably, Dobrovolsky et al are the first to provide data on the efficacy of PROTAC in vivo through experiments done in several MCL patient-derived xenograft models. PROTAC treatment resulted in tumor reduction burden as well as increased survival in a patient-derived xenograft from a patient who previously failed multiple rounds of therapy including ibrutinib. These results confirmed the in vivo value of small molecule degraders, especially for ibrutinib-refractory patients for whom few therapeutic alternatives are available.
According to a previous screening study published by the same group, BTK appeared as one of the more efficiently degradable kinase via the PROTAC platform. This technology further opens novel opportunities to selectively degrade other proteins, as recently demonstrated with bromodomain extraterminal proteins,10 making PROTACs attractive tools for both basic and clinical research. By combining the molecular effects of 2 US Food and Drug Administration–approved molecules (ie, ibrutinib and the IMiD lenalidomide), the small molecule developed by Dobrovolsky et al has an increased translational potential in B-cell lymphomas. However, several challenges remain before granting access of these novel chimeras to patients. After validation of acceptable safety as well as optimal pharmacodynamics properties for in vivo use, PROTACs will have to demonstrate a clear advantage in comparison with well-characterized targeted agents such as lenalidomide or venetoclax, both already showing promising results in combination with ibrutinib in early clinical phases.
By demonstrating the efficacy of the PROTAC strategy to counteract targeted therapy resistance in lymphoma in vivo, Dobrovolsky et al have provided an important proof of concept for the development of this new class of molecules in the future therapeutic arsenal against cancer.
Conflict-of-interest disclosure: The author declares no competing financial interests.