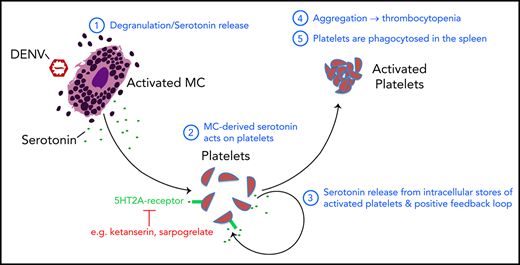
Key Points
We identified peripheral serotonin derived from mast cells to be required for dengue virus-induced thrombocytopenia.
Therapeutic targeting of platelet 5HT2 receptors reversed platelet activation, aggregation, splenic platelet uptake, and thrombocytopenia.

Abstract
Dengue virus (DENV) is the most prevalent vector-borne viral pathogen, infecting millions of patients annually. Thrombocytopenia, a reduction in circulating platelet counts, is the most consistent sign of DENV-induced disease, independent of disease severity. However, the mechanisms leading to DENV-induced thrombocytopenia are unknown. Here, we show that thrombocytopenia is caused by serotonin derived from mast cells (MCs), which are immune cells that are present in the perivascular space and are a major peripheral source of serotonin. We show that during DENV infection, MCs release serotonin, which prompts platelet activation, aggregation, and enhanced phagocytosis, dependent on 5HT2A receptors. MC deficiency in mice or pharmacologic inhibition of MCs reversed thrombocytopenia. Furthermore, reconstitution of MC-deficient mice with wild-type MCs, but not MCs lacking serotonin synthesis resulting from deficiency in the enzyme tryptophan hydroxylase-1, restored the thrombocytopenic phenotype. Exogenous serotonin was also sufficient to overcome the effects of drugs that inhibit platelet activation in vitro and to restore thrombocytopenia in DENV-infected MC-deficient mice. Therapeutic targeting of 5HT2A receptors during DENV infection effectively prevented thrombocytopenia in mice. Similarly, serotonin derived from DENV-activated human MCs led to increased human platelet activation. Thus, MC-derived serotonin is a previously unidentified mechanism of DENV-induced thrombocytopenia and a potential therapeutic target.
Introduction
Dengue virus (DENV) is an arbovirus and a member of the Flaviviridae family that causes human disease. Most infected individuals have asymptomatic or mild infection, yet some progress to severe and life-threatening illness.1 Thrombocytopenia, or reduced platelet counts, is a hallmark of DENV infection, regardless of severity.2 Although thrombocytopenia frequently occurs during mild disease, a rapid decline in platelets and accompanying bleeding is a warning sign of severe dengue. The mechanism of thrombocytopenia during dengue remains unclear, with both host and viral factors suspected to contribute.3,4
Platelets are important for hemostasis. At the end of its life or after activation, a platelet is removed through phagocytosis in the liver or spleen.5 There are 3 causes of thrombocytopenia: production defects6 ; increased turnover, as occurs in disseminated intravascular coagulation, which can be initiated by platelet binding to activated endothelium7-9 ; and sequestration, which removes platelets from the circulation.10,11 In humans, thrombocytopenia is defined as circulating fewer than 150 000 platelets per milliliter.12,13 In mice, average platelet counts are ∼1000/nL and are considered thrombocytopenia when significantly reduced.14,15 Platelet-activating mediators, including thromboxane A2 and adenosine, can be released during tissue injury or inflammation to cause thrombocytopenia.16-18 CD62P on activated platelets allows binding to monocytes.19,20 Platelet activation occurs in the blood of patients with DENV.3,21,22 Because of the role of platelets in vascular integrity,23,24 it is likely that thrombocytopenia during DENV infection promotes vascular pathology.
Mast cells (MCs) are innate immune cells densely located at the host-environmental interface, which also regulate vascular homeostasis and coagulation.25 Their granules are composed of preformed mediators, including proteases, cytokines, and monoamines. After stimulation, MCs can de novo synthesize additional mediators, including lipids and cytokines.26 These promote immune cell recruitment and activation and infection clearance, including during DENV infection.27,28 However, with widespread activation of MCs during systemic infection, MC products induce vascular leakage in animal models, and MC activation has been associated with severe dengue in humans.28-30 Whether MCs promote thrombocytopenia during dengue has not been investigated. Several studies found that MC products can activate platelets.31,32 This, coupled with evidence of platelet activation during DENV infection,3,22 led us to hypothesize that MC mediators could promote platelet activation and subsequent thrombocytopenia during DENV infection.
One mediator within MC granules that affects platelets is serotonin.33 Among immune cells, MCs are uniquely capable of synthesizing and storing serotonin and are its major source outside the central nervous system, aside from the gut enterochromaffin cells.34,35 Synthesis of this monoamine transmitter from tryptophan is controlled by the enzyme tryptophan hydroxylase (TPH), which has 2 isoforms: TPH1, produced in peripheral tissues, and TPH2, produced by neurons.36,37 The best-characterized serotonin receptor on platelets is the 5HT2A receptor, although others are present.38 Activation of platelet 5HT2A can increase adhesion molecule expression and potentiate aggregation induced by stimuli such as thrombin and collagen.39-42 Importantly, drugs targeting serotonin receptors reduce platelet activation and aggregation induced by other stimuli.43-45
Using multiple animal models of MC deficiency and DENV disease, we show that serotonin release from MCs promotes thrombocytopenia during infection. Furthermore, pharmacologic inhibition of either serotonin or MC activation prevented DENV-induced thrombocytopenia in mice or activation of human platelets in coculture. Thus, we have identified peripheral MC-derived serotonin as a previously unrecognized component of DENV-induced thrombocytopenia, revealing a potential therapeutic target of disease.
Materials and methods
Animal studies
WT (C57BL/6) mice were purchased from InVivos (Singapore). MC-deficient mice (Wsh/Wsh; ‘Sash’) and interferon (IFN) type I and type II receptor-deficient mice (IFNR-KO), both on a C57BL/6 background, were originally purchased from Jackson Laboratories and bred in-house. Mcpt5-DTA mice were generated by crossing Mcpt5-Cre mice (from Axel Roers, Heidelberg University) to a DTA reporter line (Jackson Laboratories).
Ethical approvals
For studies using human blood, approvals were obtained from the National University of Singapore institutional review board, and donors provided written informed consent. Animal experiments were performed according to protocols approved by the SingHealth Institutional Animal Care and Use Committee.
Additional methods are provided in the supplemental Materials, available on the Blood Web site.
Results
DENV-induced thrombocytopenia is caused by platelet destruction in the spleen
Platelet activation is present in the blood of patients with dengue.21,22 However, data in experimental animal models replicating this phenomenon are lacking, despite demonstrating thrombocytopenia.46,47 We used 2 models of DENV infection: a wild-type (WT) immune-competent model that experiences lower levels of virus replication, and an IFNR-KO model, which is commonly used to study viral infections, as they exhibit high levels of viremia and death. In both models, DENV infection results in thrombocytopenia. In WT mice, infected intraperitoneally with 1 × 106 plaque-forming units (PFU) of DENV2, thrombocytopenia started 1 day postinfection, with recovery in platelet counts at 6 days (Figure 1A). DENV infection with 2 × 105 PFU was lethal in the IFNR-KO model, and thrombocytopenia was also observed (supplemental Figure 1). In both models, platelets of the DENV-infected animals were activated, indicated by an increase in platelets expressing activation marker CD62P+ (Figure 1B; supplemental Figure 1C; gating strategy in supplemental Figure 2A). Activated platelets were also increased in the spleens of DENV-infected mice (Figure 1C), and increased platelet aggregates were observed in the blood (Figure 1D). Thus, our findings demonstrate that both mouse models display thrombocytopenia and platelet activation, similar to observations during clinical dengue.
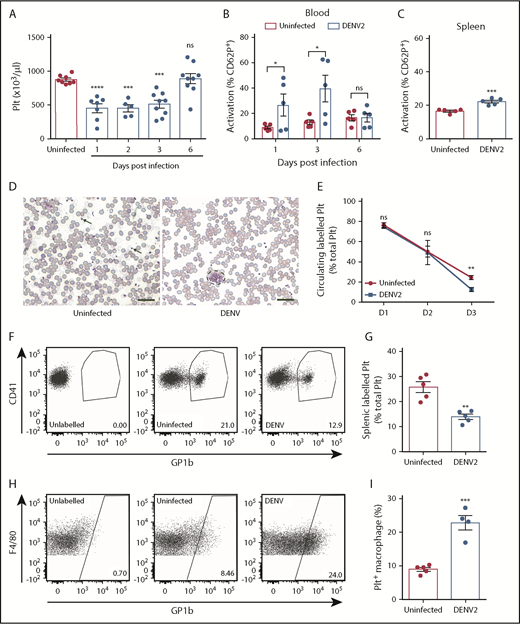
DENV infection results in thrombocytopenia because of increased platelet activation and uptake. (A) Platelet counts of C57Bl/6 mice infected with DENV2 (1 × 106 PFU via the intraperitoneal route; n = 5-9 per group). Blood and spleens of DENV2-infected mice were isolated, and cells were stained for platelet marker CD41 and activation marker CD62P and analyzed by flow cytometry (n = 5 per group). (B) There was increased activation of platelets in the blood at days 1 and 3 postinfection, with resolution at day 6. (C) Increased platelet activation in the spleen was observed 72 hours postinfection. Flow cytometry gating strategy is presented in supplemental Figure 2A. (D) Representative eosin and methylene blue staining of peripheral blood smears 24 hours postinfection. Some individual platelets are indicated by arrows. Platelet aggregates are surrounded by a dashed line. Scale bar, 25 μm. (E) Platelets were labeled in vivo to track their turnover by injecting mice with an antibody against platelet GP1b before DENV infection. Blood was taken at indicated points, stained for CD41, and analyzed by flow cytometry (n = 5 per group). Significantly reduced labeled platelets were observed in DENV-infected mice compared with uninfected control mice at 72 hours postinfection, indicating platelet destruction. (F) Representative flow cytometry plots of labeled circulating platelets 3 days postinfection. (G-I) Spleens were harvested from DENV2-infected mice 3 days postinfection; stained for CD41, CD11b, and F4/80; and analyzed by flow cytometry (n = 5 per group). (G) A significant reduction in total splenic labeled platelets was observed during DENV infection. (H) Representative flow diagram for macrophages containing platelets (CD11b+F4/80+GP1b+). Flow cytometry gating strategy is presented in supplemental Figure 2B. (I) Significantly increased numbers of macrophages had phagocytosed platelets (CD11b+F4/80+GP1b+) in spleens of DENV-infected mice compared with control, uninfected mice (n = 4-5 per group). Error bars represent the standard error of the mean. P values were determined by 1-way ANOVA for panel A, or by Student’s unpaired t test for all other panels, and error bars represent the standard error of the mean. ns, not significant; *P < .05; **P < .01; ***P < .001; ****P < .0001.
DENV infection results in thrombocytopenia because of increased platelet activation and uptake. (A) Platelet counts of C57Bl/6 mice infected with DENV2 (1 × 106 PFU via the intraperitoneal route; n = 5-9 per group). Blood and spleens of DENV2-infected mice were isolated, and cells were stained for platelet marker CD41 and activation marker CD62P and analyzed by flow cytometry (n = 5 per group). (B) There was increased activation of platelets in the blood at days 1 and 3 postinfection, with resolution at day 6. (C) Increased platelet activation in the spleen was observed 72 hours postinfection. Flow cytometry gating strategy is presented in supplemental Figure 2A. (D) Representative eosin and methylene blue staining of peripheral blood smears 24 hours postinfection. Some individual platelets are indicated by arrows. Platelet aggregates are surrounded by a dashed line. Scale bar, 25 μm. (E) Platelets were labeled in vivo to track their turnover by injecting mice with an antibody against platelet GP1b before DENV infection. Blood was taken at indicated points, stained for CD41, and analyzed by flow cytometry (n = 5 per group). Significantly reduced labeled platelets were observed in DENV-infected mice compared with uninfected control mice at 72 hours postinfection, indicating platelet destruction. (F) Representative flow cytometry plots of labeled circulating platelets 3 days postinfection. (G-I) Spleens were harvested from DENV2-infected mice 3 days postinfection; stained for CD41, CD11b, and F4/80; and analyzed by flow cytometry (n = 5 per group). (G) A significant reduction in total splenic labeled platelets was observed during DENV infection. (H) Representative flow diagram for macrophages containing platelets (CD11b+F4/80+GP1b+). Flow cytometry gating strategy is presented in supplemental Figure 2B. (I) Significantly increased numbers of macrophages had phagocytosed platelets (CD11b+F4/80+GP1b+) in spleens of DENV-infected mice compared with control, uninfected mice (n = 4-5 per group). Error bars represent the standard error of the mean. P values were determined by 1-way ANOVA for panel A, or by Student’s unpaired t test for all other panels, and error bars represent the standard error of the mean. ns, not significant; *P < .05; **P < .01; ***P < .001; ****P < .0001.
To determine whether thrombocytopenia in DENV-infected animals was the result of a platelet production defect, sequestration in the spleen, or increased turnover, we used an established protocol to label the platelets of mice in vivo before DENV infection.48 There was a reduction in the proportion of labeled circulating platelets in DENV-infected animals (Figure 1E-F), significant by 3 days postinfection, which is consistent with their removal at time points after activation during DENV infection. The spleen is a major site of platelet sequestration, and splenic phagocytes can remove activated or senesced platelets.49,50 Therefore, we examined the spleen for evidence of enhanced uptake of labeled platelets within splenic macrophages (CD11b+F4/80+) by flow cytometry (gating strategy in supplemental Figure 2B). In DENV-infected mice, total numbers of free-labeled platelets in the spleen were reduced compared with uninfected controls (Figure 1G), suggesting against sequestration. Conversely, the proportion of splenic macrophages that were positive for labeled platelets increased (Figure 1H), suggesting that more splenic macrophages had taken up labeled platelets during DENV infection than at steady state (Figure 1I). Together, these data show that platelet destruction, resulting from activation and subsequent phagocytosis in the spleen, underlies the thrombocytopenic phenotype seen in DENV infection.
MCs promote thrombocytopenia by increasing platelet activation and uptake
To investigate whether MCs also contribute to thrombocytopenia, we infected WT and MC-deficient (Sash) mice with DENV. In contrast to WT mice, which experienced thrombocytopenia by 24 hours postinfection (Figure 1A), there was not a significant decrease in platelets in DENV-infected Sash mice, even at 3 days postinfection (Figure 2A). Activation of circulating platelets also did not increase (Figure 2B), nor did platelet uptake by splenic phagocytes (Figure 2C) in DENV-infected Sash mice compared with uninfected controls. This was in spite of previous studies showing that viral titers in Sash mice were moderately higher compared with WT mice.27 To determine whether MCs were sufficient to induce thrombocytopenia, Sash mice were reconstituted (Sash-R) with bone marrow derived mast cells (BMMCs). Reconstitution restored thrombocytopenia (Figure 2D), which was characterized by increased platelet activation (Figure 2E) and increased platelet uptake by splenic macrophages (Figure 2F). Platelet aggregates were observed in peripheral blood films of WT and Sash-R mice, but not DENV-infected Sash mice (Figure 2G), supporting the requirement of MCs for DENV-induced platelet activation and aggregation. We confirmed this using a second model of MC deficiency, the Mcpt5-cre+DTA+ model.51 MC-deficient Mcpt5-cre+DTA+ mice and their MC-sufficient DTA+ littermates were infected with DENV2. Infection of mice resulted in thrombocytopenia (supplemental Figure 3A) and increased platelet activation in MC-sufficient DTA+, but not MC-deficient Mcpt5-cre+DTA+ mice (supplemental Figure 3B). This was also reflected in the peripheral blood, where platelet aggregates were seen in DTA+ mice but not in Mcpt5-cre+DTA+ mice (supplemental Figure 3C). Thus, consistent results were obtained in 2 mouse models of MC deficiency that MCs promote DENV-induced platelet activation and aggregation.
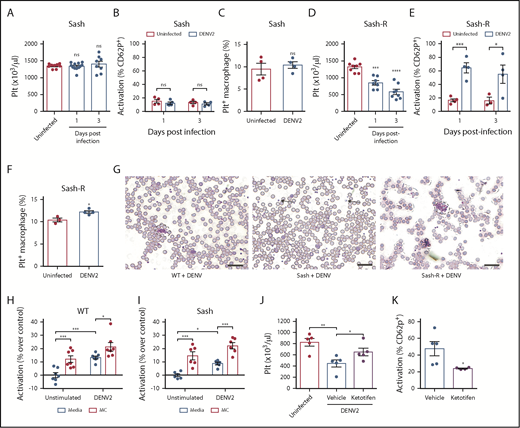
DENV-induced thrombocytopenia is MC-dependent. (A) Platelet counts were not reduced in MC-deficient (Sash) mice infected with DENV2 (1 × 106 PFU via intraperitoneal injection) at 24 and 72 hours postinfection (n = 8-12 per group). (B) No significant differences in platelet activation were observed between uninfected and DENV2-infected Sash mice, determined by staining blood isolated 24 and 72 hours postinfection for platelet marker CD41 and activation marker CD62P, followed by analysis by flow cytometry (n = 4-5 per group). Flow cytometry gating strategy is presented in supplemental Figure 2A. (C) Platelets were labeled in mice through injection of an antibody against platelet GP1b. Spleens of DENV2-infected and control Sash mice were isolated 3 days postinfection, and cells were stained for CD11b and F4/80 and analyzed by flow cytometry. No significant differences in the numbers of Platelet+ Macrophages (CD11b+F4/80+GP1b+) were observed between uninfected and DENV-infected Sash mice (n = 4). Flow cytometry gating strategy is presented in supplemental Figure 2B. (D) Sash mice that were reconstituted with MCs (Sash-R) were infected with DENV2. A significant reduction in circulating platelets was observed in Sash-R mice infected with DENV compared with uninfected controls at 24 and 72 hours postinfection (n = 7-8 per group). (E) Increased numbers of activated platelets (CD41+CD62P+) were detected in the circulation of DENV-infected Sash-R mice compared with uninfected controls at 24 and 72 hours postinfection (n = 3-4 per group). (F) Increased numbers of Platelet+ Macrophages were detected in the spleens of DENV-infected Sash-R mice compared with uninfected control spleens (n = 3-4 per group). (G) Representative eosin and methylene blue staining of peripheral blood smears from WT, Sash and Sash-R mice, day 3 postinfection showing aggregates in infected WT and Sash-R mice, but not Sash mice. Some individual platelets are indicated by arrows. Platelet aggregates are surrounded by dashed lines. Scale bar, 25 μm. Whole blood from (H) WT or (I) Sash mice was isolated and stimulated with DENV2, or cocultured with MCs or MCs+DENV2 for 15 minutes. Cells were then fixed and stained for CD41 and CD62P and analyzed by flow cytometry (n = 6-7 per group). For panels D-E, coculture with DENV or MCs raised the activation levels of platelets, but was significantly enhanced with coculture with MCs+DENV2. (J) Ketotifen treatment (6 mg/kg) of DENV-infected mice significantly increased platelet counts compared with vehicle treatment (24 hours postinfection). (K) Ketotifen also reduced platelet activation compared with controls receiving vehicle treatment (n = 4-5 per group). Error bars represent the standard error of the mean. P values were determined by Student’s unpaired t test. ns, not significant; *P < .05; **P < .01; ***P < .001; ****P < .0001.
DENV-induced thrombocytopenia is MC-dependent. (A) Platelet counts were not reduced in MC-deficient (Sash) mice infected with DENV2 (1 × 106 PFU via intraperitoneal injection) at 24 and 72 hours postinfection (n = 8-12 per group). (B) No significant differences in platelet activation were observed between uninfected and DENV2-infected Sash mice, determined by staining blood isolated 24 and 72 hours postinfection for platelet marker CD41 and activation marker CD62P, followed by analysis by flow cytometry (n = 4-5 per group). Flow cytometry gating strategy is presented in supplemental Figure 2A. (C) Platelets were labeled in mice through injection of an antibody against platelet GP1b. Spleens of DENV2-infected and control Sash mice were isolated 3 days postinfection, and cells were stained for CD11b and F4/80 and analyzed by flow cytometry. No significant differences in the numbers of Platelet+ Macrophages (CD11b+F4/80+GP1b+) were observed between uninfected and DENV-infected Sash mice (n = 4). Flow cytometry gating strategy is presented in supplemental Figure 2B. (D) Sash mice that were reconstituted with MCs (Sash-R) were infected with DENV2. A significant reduction in circulating platelets was observed in Sash-R mice infected with DENV compared with uninfected controls at 24 and 72 hours postinfection (n = 7-8 per group). (E) Increased numbers of activated platelets (CD41+CD62P+) were detected in the circulation of DENV-infected Sash-R mice compared with uninfected controls at 24 and 72 hours postinfection (n = 3-4 per group). (F) Increased numbers of Platelet+ Macrophages were detected in the spleens of DENV-infected Sash-R mice compared with uninfected control spleens (n = 3-4 per group). (G) Representative eosin and methylene blue staining of peripheral blood smears from WT, Sash and Sash-R mice, day 3 postinfection showing aggregates in infected WT and Sash-R mice, but not Sash mice. Some individual platelets are indicated by arrows. Platelet aggregates are surrounded by dashed lines. Scale bar, 25 μm. Whole blood from (H) WT or (I) Sash mice was isolated and stimulated with DENV2, or cocultured with MCs or MCs+DENV2 for 15 minutes. Cells were then fixed and stained for CD41 and CD62P and analyzed by flow cytometry (n = 6-7 per group). For panels D-E, coculture with DENV or MCs raised the activation levels of platelets, but was significantly enhanced with coculture with MCs+DENV2. (J) Ketotifen treatment (6 mg/kg) of DENV-infected mice significantly increased platelet counts compared with vehicle treatment (24 hours postinfection). (K) Ketotifen also reduced platelet activation compared with controls receiving vehicle treatment (n = 4-5 per group). Error bars represent the standard error of the mean. P values were determined by Student’s unpaired t test. ns, not significant; *P < .05; **P < .01; ***P < .001; ****P < .0001.
To confirm that there was no intrinsic deficit in the ability of platelets of MC-deficient mice to be activated, we stimulated platelets ex vivo with media containing DENV, supernatants from unstimulated MCs or supernatants of DENV-stimulated MCs, or control media. Platelets from both WT (Figure 2H) and Sash (Figure 2I) mice could be stimulated by DENV-elicited MC products, resulting in increased CD62P+ platelets. Administration of ketotifen, a MC stabilizer that blocks DENV-induced MC degranulation,28,52 significantly reduced thrombocytopenia (Figure 2J) and platelet activation (Figure 2K) during DENV infection. Thus, both genetic and pharmacologic inhibition approaches confirm that the release of products from MCs during DENV infection promotes thrombocytopenia.
MCs promote thrombocytopenia through serotonin release during DENV infection
We next aimed to identify the MC-derived mediators responsible for DENV-induced platelet activation. Because platelets express receptors for serotonin, we questioned whether serotonin was released by DENV-activated MCs. Both human and rodent MCs produce serotonin.34 DENV stimulation of MCs induced serotonin release that could be observed visually in vitro, coinciding with degranulation (Figure 3A) and measured quantitatively (Figure 3B). Unlike in WT mice, DENV infection did not increase plasma serotonin levels in Sash mice (Figure 3C). To demonstrate in vivo the contribution of MC serotonin to DENV-induced thrombocytopenia, we generated mice selectively deficient in MC serotonin through reconstitution of Sash mice with BMMCs deficient in TPH-1 (TPH1-KO). (Schematic provided in supplemental Figure 4A.) TPH1-KO BMMCs were generated using a CRISPR/Cas9 method and validated to have significantly reduced serotonin production (supplemental Figure 4B-C). Mice lacking MC serotonin (Sash-RTPH1-KO) had no differences in resting platelet counts (supplemental Figure 4D) or activation status (supplemental Figure 4E) compared with Sash mice that were reconstituted with WT BMMCs (Sash-RWT). However, the Sash-RTPH1-KO group had reduced thrombocytopenia compared with Sash-RWT after DENV infection (Figure 3D). Mice lacking MC serotonin had reduced platelet activation (Figure 3E), despite comparable levels of MC degranulation (supplemental Figure 4F) and infection (supplemental Figure 4G). We observed platelet aggregates in the blood of DENV-infected Sash-RWT mice, but not Sash-RTPH1-KO mice (Figure 3F). To test whether serotonin was sufficient to reverse this phenotype, we infected Sash mice with DENV and treated them with exogenous serotonin, which restored thrombocytopenia (Figure 3G) and platelet activation (Figure 3H). These results support that MCs promote thrombocytopenia through the release of serotonin during DENV infection.
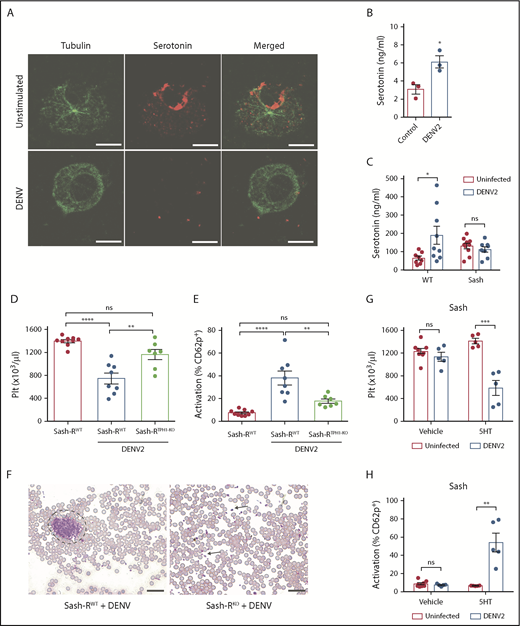
MC-derived serotonin promotes DENV-induced thrombocytopenia. (A) Serotonin was detected by immunostaining of control BMMCs, whereas activation of BMMCs and release of serotonin was observed after incubation with DENV2 at a multiplicity of infection of 1 for 2 hours. Scale bar, 20 μm. (B) BMMCs release serotonin into the cell culture supernatant after DENV exposure as in panel A, measured by enzyme-linked immunosorbent assay. (C) WT and Sash mice were infected with DENV2 (1 × 106 PFU via intraperitoneal injection). At 24 hours postinfection, plasma was isolated and serotonin levels were measured by enzyme-linked immunosorbent assay. (D-F) Mice selectively deficient in MC serotonin were generated through reconstitution of Sash mice with BMMCs deficient in TPH-1 (Sash-RTPH1-KO). For controls, mice were reconstituted with BMMCs from WT mice (Sash-RWT). A schematic of the experimental strategy is provided in supplemental Figure 4A. (D) Platelet counts decreased significantly in Sash-RWT infected with DENV2, but not in infected Sash-RTPH1-KO mice at day 3 postinfection (n = 4-7 per group). (E) Increased numbers of activated platelets (CD41+CD62P+) were detected in the circulation of DENV-infected Sash-RWT mice, but not in infected Sash-RTPH1-KO compared with uninfected controls. Flow cytometry gating strategy is presented in supplemental Figure 2A (n = 7-9 per group). (F) Representative eosin and methylene blue staining of peripheral blood smears day 3 postinfection. Some individual platelets are indicated by arrows. Platelet aggregates are surrounded by a dashed line. Scale bar, 25 μm. (G) Sash mice were infected with DENV2 and treated with 150 mg/kg serotonin or vehicle daily. Blood was taken for analysis 3 days postinfection. In infected animals, treatment with serotonin significantly reduced circulating platelet counts compared with vehicle treatment zanimals. (H) Serotonin treatment resulted in increased platelet activation in infected mice compared with vehicle-treated controls (n = 5-8 per group). Means are presented with error bars representing the standard error of the mean. P values were determined by 1-way ANOVA for panels C-D or by Student’s unpaired t test for panels B, F-G. ns, not significant; *P < .05; **P < .01; ***P < .001; ****P < .0001.
MC-derived serotonin promotes DENV-induced thrombocytopenia. (A) Serotonin was detected by immunostaining of control BMMCs, whereas activation of BMMCs and release of serotonin was observed after incubation with DENV2 at a multiplicity of infection of 1 for 2 hours. Scale bar, 20 μm. (B) BMMCs release serotonin into the cell culture supernatant after DENV exposure as in panel A, measured by enzyme-linked immunosorbent assay. (C) WT and Sash mice were infected with DENV2 (1 × 106 PFU via intraperitoneal injection). At 24 hours postinfection, plasma was isolated and serotonin levels were measured by enzyme-linked immunosorbent assay. (D-F) Mice selectively deficient in MC serotonin were generated through reconstitution of Sash mice with BMMCs deficient in TPH-1 (Sash-RTPH1-KO). For controls, mice were reconstituted with BMMCs from WT mice (Sash-RWT). A schematic of the experimental strategy is provided in supplemental Figure 4A. (D) Platelet counts decreased significantly in Sash-RWT infected with DENV2, but not in infected Sash-RTPH1-KO mice at day 3 postinfection (n = 4-7 per group). (E) Increased numbers of activated platelets (CD41+CD62P+) were detected in the circulation of DENV-infected Sash-RWT mice, but not in infected Sash-RTPH1-KO compared with uninfected controls. Flow cytometry gating strategy is presented in supplemental Figure 2A (n = 7-9 per group). (F) Representative eosin and methylene blue staining of peripheral blood smears day 3 postinfection. Some individual platelets are indicated by arrows. Platelet aggregates are surrounded by a dashed line. Scale bar, 25 μm. (G) Sash mice were infected with DENV2 and treated with 150 mg/kg serotonin or vehicle daily. Blood was taken for analysis 3 days postinfection. In infected animals, treatment with serotonin significantly reduced circulating platelet counts compared with vehicle treatment zanimals. (H) Serotonin treatment resulted in increased platelet activation in infected mice compared with vehicle-treated controls (n = 5-8 per group). Means are presented with error bars representing the standard error of the mean. P values were determined by 1-way ANOVA for panels C-D or by Student’s unpaired t test for panels B, F-G. ns, not significant; *P < .05; **P < .01; ***P < .001; ****P < .0001.
MC serotonin increases platelet activation to DENV via 5HT2 receptors
To determine how serotonin affected platelets during DENV infection, we stimulated mouse platelets with DENV and serotonin ex vivo. Serotonin itself both induced platelet activation and also further increased DENV-induced platelet activation in a dose-dependent manner (Figure 4A). Although either serotonin or DENV could directly activate platelets in vitro, neither induced platelet activation or thrombocytopenia in vivo independently (Figure 3G-H). To reconcile these observations and define the contribution of serotonin to thrombocytopenia during DENV infection, we infected WT mice with DENV and administered exogenous serotonin, hypothesizing that the increase in serotonin-mediated platelet activation would worsen thrombocytopenia in vivo. Consistent with in vitro observations (Figure 4A), we observed lower platelet counts in serotonin-treated DENV-infected mice (Figure 4B) and increased platelet activation (Figure 4C) relative to vehicle-treated mice. Similarly, platelet aggregates increased in the blood of serotonin-treated mice (Figure 4D). As we observed that MC serotonin increased the responsiveness of platelets to DENV (Figures 2H and 3D-E), we questioned whether serotonin acted on the major serotonin receptor on platelets, 5HT2A.53,54 We stimulated mouse platelets with BMMCs and DENV in the presence of the 5HT2A antagonist, ketanserin. Blockade of 5HT2A abolished the increased platelet activation induced by DENV-stimulated MC-derived mediators (Figure 4E), indicating that the action of MC-derived serotonin on platelets is via the 5HT2A receptor.
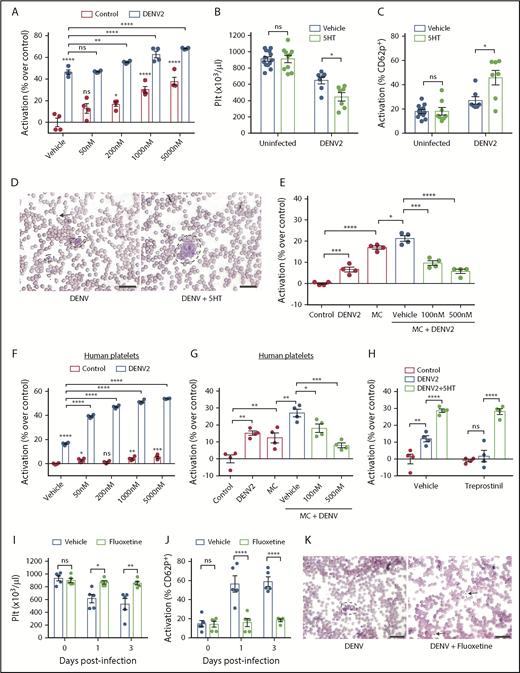
Serotonin exacerbates platelet activation to DENV via 5HT2 receptors and downstream pathways. (A) Whole blood from WT mice was isolated and stimulated ex vivo with DENV2, in the presence of serotonin (50 nM, 200 nM, 1 μM or 5 μM) for 15 minutes. Cells were then fixed and stained for platelet marker CD41 and activation marker CD62P and analyzed by flow cytometry (n = 4). The flow cytometry gating strategy is presented in supplemental Figure 2A. DENV raised the activation levels of platelets, which was significantly further increased in the presence of serotonin. (B) WT mice were infected with DENV2 (1 × 106 PFU I) and treated with 150 mg/kg of serotonin or vehicle daily. Serotonin treatment worsened thrombocytopenia during DENV infection compared with vehicle-treated mice, at 3 days postinfection. (C) DENV infection resulted in increased platelet activation, which was further increased when mice were also treated with exogenous serotonin (n = 7-13 per group). (D) Representative peripheral blood smears, day 3 postinfection, show platelet aggregation (dashed lines) was more visually apparent after exogenous serotonin treatment compared with DENV infection. Some individual platelets are indicated by arrows. Scale bar = 25 μm. (E) Whole blood from WT mice was isolated and stimulated with DENV2, MCs or MC+DENV2 in the presence of vehicle or ketanserin (100 nM or 500 nM) for 15 minutes. Cells were then analyzed by flow cytometry. Coculture with DENV or MCs raised the activation levels of platelets but was significantly further enhanced in the presence of MC+DENV2. This was abolished with ketanserin treatment (n = 4). (F) Whole blood from healthy human volunteers was isolated and stimulated with DENV2, serotonin, or DENV2+serotonin for 15 minutes. Cells were then fixed and stained for platelet markers CD41 and CD62P and analyzed by flow cytometry. DENV raised the activation levels of platelets. DENV in the presence of serotonin significantly enhanced platelet activation over control and DENV groups (n = 4). (G) Human whole blood was stimulated with DENV2, MCs or DENV2+MCs in the presence of vehicle or ketanserin (100 nM or 500 nM) for 15 minutes before assessment of platelet activation. Ketanserin treatment abolished the significant increase in platelet activation that occurred in the DENV2+MC treated group compared with controls (n = 4). (H) Mouse blood was incubated with 25 nM treprostinil for 10 minutes before stimulation with DENV and 5 μM serotonin for 15 minutes, followed by assessment of platelet activation. Treprostinil prevented DENV-induced platelet activation, which was overcome in the presence of serotonin (n = 4). (I-K) WT mice (n = 5 per group) were treated with either fluoxetine or vehicle for 2 weeks and subsequently infected with DENV2 (1 × 106 PFU intraperitoneally). (I) Fluoxetine pretreatment prevented thrombocytopenia after DENV2 infection. (J) There was reduced platelet activation when mice were treated with fluoxetine prior to DENV2 infection. (K) Representative peripheral blood smears, day 3 postinfection, show decreased platelet aggregation (dashed lines) when mice were treated with fluoxetine. Some individual platelets are indicated by arrows. Scale bar = 25 μm. Error bars represent the standard error of the mean. P values were determined by 1-way ANOVA or by Student’s unpaired t test for panels in vivo experiments. ns, not significant; *P < .05; **P < .01; ***P < .001; ****P < .0001.
Serotonin exacerbates platelet activation to DENV via 5HT2 receptors and downstream pathways. (A) Whole blood from WT mice was isolated and stimulated ex vivo with DENV2, in the presence of serotonin (50 nM, 200 nM, 1 μM or 5 μM) for 15 minutes. Cells were then fixed and stained for platelet marker CD41 and activation marker CD62P and analyzed by flow cytometry (n = 4). The flow cytometry gating strategy is presented in supplemental Figure 2A. DENV raised the activation levels of platelets, which was significantly further increased in the presence of serotonin. (B) WT mice were infected with DENV2 (1 × 106 PFU I) and treated with 150 mg/kg of serotonin or vehicle daily. Serotonin treatment worsened thrombocytopenia during DENV infection compared with vehicle-treated mice, at 3 days postinfection. (C) DENV infection resulted in increased platelet activation, which was further increased when mice were also treated with exogenous serotonin (n = 7-13 per group). (D) Representative peripheral blood smears, day 3 postinfection, show platelet aggregation (dashed lines) was more visually apparent after exogenous serotonin treatment compared with DENV infection. Some individual platelets are indicated by arrows. Scale bar = 25 μm. (E) Whole blood from WT mice was isolated and stimulated with DENV2, MCs or MC+DENV2 in the presence of vehicle or ketanserin (100 nM or 500 nM) for 15 minutes. Cells were then analyzed by flow cytometry. Coculture with DENV or MCs raised the activation levels of platelets but was significantly further enhanced in the presence of MC+DENV2. This was abolished with ketanserin treatment (n = 4). (F) Whole blood from healthy human volunteers was isolated and stimulated with DENV2, serotonin, or DENV2+serotonin for 15 minutes. Cells were then fixed and stained for platelet markers CD41 and CD62P and analyzed by flow cytometry. DENV raised the activation levels of platelets. DENV in the presence of serotonin significantly enhanced platelet activation over control and DENV groups (n = 4). (G) Human whole blood was stimulated with DENV2, MCs or DENV2+MCs in the presence of vehicle or ketanserin (100 nM or 500 nM) for 15 minutes before assessment of platelet activation. Ketanserin treatment abolished the significant increase in platelet activation that occurred in the DENV2+MC treated group compared with controls (n = 4). (H) Mouse blood was incubated with 25 nM treprostinil for 10 minutes before stimulation with DENV and 5 μM serotonin for 15 minutes, followed by assessment of platelet activation. Treprostinil prevented DENV-induced platelet activation, which was overcome in the presence of serotonin (n = 4). (I-K) WT mice (n = 5 per group) were treated with either fluoxetine or vehicle for 2 weeks and subsequently infected with DENV2 (1 × 106 PFU intraperitoneally). (I) Fluoxetine pretreatment prevented thrombocytopenia after DENV2 infection. (J) There was reduced platelet activation when mice were treated with fluoxetine prior to DENV2 infection. (K) Representative peripheral blood smears, day 3 postinfection, show decreased platelet aggregation (dashed lines) when mice were treated with fluoxetine. Some individual platelets are indicated by arrows. Scale bar = 25 μm. Error bars represent the standard error of the mean. P values were determined by 1-way ANOVA or by Student’s unpaired t test for panels in vivo experiments. ns, not significant; *P < .05; **P < .01; ***P < .001; ****P < .0001.
To confirm that human MCs activate platelets through 5HT2A, we cocultured human platelets with serotonin, starting at concentrations consistent with those measured in healthy human plasma.55,56 This resulted in increased platelet activation (Figure 4F). In addition, the enhanced activation in response to DENV was dependent on the 5HT2A receptor, as the effect was abolished by ketanserin (Figure 4G). Thus, MC serotonin triggers human platelet activation in response to DENV via the 5HT2A receptor.
Although augmented by serotonin, DENV was sufficient to induce platelet activation in vitro (Figure 4A,E-G), whereas in vivo, DENV was insufficient to induce thrombocytopenia in the absence of MC serotonin (Figures 2A and 3G). Because blood contains inhibitory factors, such as prostacyclin, that limit platelet activation and aggregation in vivo,57,58 we hypothesized that MC-derived serotonin acted to overcome inhibitory factors during DENV infection, leading to thrombocytopenia. To address this, we treated mouse platelets with the prostacyclin analog treprostinil before stimulation with DENV. We observed that DENV did not cause platelet activation in the presence of trepostinil (Figure 4H). However, with the addition of serotonin and DENV, significant platelet activation was observed in the presence of treprostinil (Figure 4H). Our results demonstrate that both DENV and serotonin are required for thrombocytopenia in vivo, and that plasma platelet inhibitors are a likely explanation for why DENV is sufficient to induce platelet activation in the absence of serotonin in vitro, but not in vivo.
Platelets also store large amounts of circulating serotonin in their dense granules, and release them on activation. Because MC-derived serotonin promotes thrombocytopenia (Figure 3D), we questioned whether serotonin release subsequent to platelet activation would further augment DENV-induced thrombocytopenia. For this, we depleted platelet serotonin in WT mice through the established method of chronic fluoxetine treatment.59,60 After verifying platelet serotonin depletion (supplemental Figure 5A), we subsequently infected the mice with DENV. Fluoxetine pretreatment alone did not affect platelet counts, but prevented DENV-induced thrombocytopenia (Figure 4I). Consistent with this, neither uninfected nor DENV-infected fluoxetine-treated mice had increased platelet activation (Figure 4J). Fluoxetine pretreated DENV-infected mice also showed no platelet aggregates in blood, unlike untreated mice (Figure 4K). Because fluoxetine has been reported to reduce DENV replication in cell culture,61 we also checked but found no difference in the infection levels in the spleen (supplemental Figure 5B), suggesting that the effects of fluoxetine on platelet serotonin were responsible for the observed phenotype. Fluoxetine treatment also did not affect vascular leakage after DENV infection (supplemental Figure 5C). Thus, after initiation by MC-derived serotonin, the release of serotonin from activated platelets contributes to thrombocytopenia in a positive feedback loop during DENV infection.
Our data show that MC serotonin can activate platelets using 5HT2A receptors, which is compounded by release of stored serotonin from platelets; however, the binding of serotonin to GTPases, or serotonylation, has recently been described to contribute to platelet activation.62 To determine whether serotonylation is activated during DENV infection, we stimulated platelets from WT mice with serotonin and DENV in the presence of the serotonylation inhibitor cystamine. Indeed, inhibition of serotonylation reduced the levels of platelet activation (supplemental Figure 6A). Similarly, using human platelets, inhibition of serotonylation also reduced platelet activation in response to combined DENV and serotonin stimulation (supplemental Figure 6B), showing that serotonin induces multiple platelet activation pathways.
5HT2 receptor antagonism therapeutically reverses DENV-induced thrombocytopenia
Of the multiple serotonin-dependent pathways that are activated as a result of MC-derived serotonin during DENV infection, 5HT2 receptors are an upstream target that were able to entirely abrogate the downstream thrombocytopenic response in vivo. Several drugs that act as antagonists of the 5HT2A receptor have been approved for clinical use. To determine whether they could reduce DENV-induced thrombocytopenia, we infected WT mice with DENV and treated them with ketanserin. Ketanserin treatment rescued thrombocytopenia (Figure 5A), reduced platelet activation (Figure 5B), reduced platelet uptake by splenic macrophages (Figure 5C), and reduced platelet aggregates in the blood (Figure 5D). We next confirmed our findings in the second model of severe DENV infection, using IFNR-KO mice. Similarly, treatment with ketanserin reduced thrombocytopenia (Figure 5E) and platelet activation (Figure 5F) during DENV infection in IFNR-KO mice. Because inhibition of host factors may affect viral replication, we measured levels of viremia after DENV infection. There was no significant difference in serum DENV genome copy numbers in ketanserin-treated mice in both models (supplemental Figure 7A-B). We also measured mRNA levels of interleukin 6 and interleukin 10 in the spleen, as these 2 cytokines are known to be increased in DENV infection.63 Ketanserin treatment did not affect vascular leakage (supplemental Figure 7C), viremia, or levels of either cytokine in the spleen (supplemental Figure 7D-F). To further support that 5HT2A receptor targeting is a viable strategy to prevent DENV-induced thrombocytopenia, we used another clinically available 5HT2A receptor antagonist, sarpogrelate. Sarpogrelate rescued thrombocytopenia during DENV infection (Figure 5G). These results highlight that inhibition of 5HT2A receptors represents a viable therapeutic strategy in the treatment of dengue.
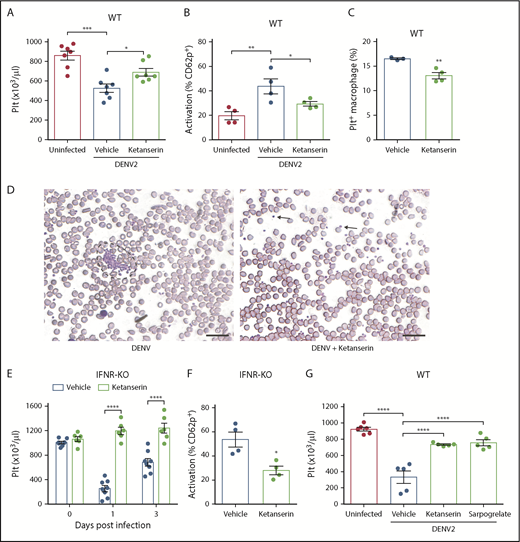
Targeting serotonin receptors reduces thrombocytopenia by limiting platelet activation. (A) DENV infection in WT mice decreased platelet counts 3 days postinfection, which was reversed by ketanserin treatment (8 mg/kg; n = 7). (B) Ketanserin treatment in DENV-infected mice significantly reduced platelet activation compared with vehicle-treated controls, determined by flow cytometry, after staining blood isolated 24 hours postinfection for activated platelets (CD41+CD62P+) (n = 4). Flow cytometry gating strategy is presented in supplemental Figure 2A. (C) Platelets were labeled in mice through injection of an antibody against GP1b before infection. Spleens of DENV2-infected mice were isolated 3 days postinfection, and cells were stained for CD11b and F4/80 and analyzed by flow cytometry. There was a significant decrease in the numbers of Platelet+ Macrophages (CD11b+F4/80+GP1b+) in ketanserin-treated mice compared with vehicle-treated controls (n = 3-4). Flow cytometry gating strategy is presented in supplemental Figure 2B. (D) Staining of peripheral blood smears day 3 postinfection showed the presence of aggregates (surrounded by dashed lines) in vehicle-treated mice but no aggregates (individual platelets indicated by arrow) in ketanserin-treated mice. Scale bar, 25 μm. (E-F) IFNR-KO mice were infected with DENV2 and treated with 8 mg/kg ketanserin or vehicle daily. (E) Ketanserin treatment rescued platelet counts during DENV infection. (F) Ketanserin treatment of DENV-infected mice resulted in a significant reduction in platelet activation compared with vehicle treated-controls at day 3 postinfection (n = 4-5 per group). (G) WT mice were infected with DENV2 and treated with ketanserin (8 mg/kg), sarpogrelate (3 mg/kg), or vehicle. Treatment with either 5HT2A antagonist rescued thrombocytopenia during DENV infection, compared with vehicle-treated controls. Data represent the mean and error bars represent the standard error of the mean. P values were determined by Student’s unpaired t test or by 1-way ANOVA (G). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Targeting serotonin receptors reduces thrombocytopenia by limiting platelet activation. (A) DENV infection in WT mice decreased platelet counts 3 days postinfection, which was reversed by ketanserin treatment (8 mg/kg; n = 7). (B) Ketanserin treatment in DENV-infected mice significantly reduced platelet activation compared with vehicle-treated controls, determined by flow cytometry, after staining blood isolated 24 hours postinfection for activated platelets (CD41+CD62P+) (n = 4). Flow cytometry gating strategy is presented in supplemental Figure 2A. (C) Platelets were labeled in mice through injection of an antibody against GP1b before infection. Spleens of DENV2-infected mice were isolated 3 days postinfection, and cells were stained for CD11b and F4/80 and analyzed by flow cytometry. There was a significant decrease in the numbers of Platelet+ Macrophages (CD11b+F4/80+GP1b+) in ketanserin-treated mice compared with vehicle-treated controls (n = 3-4). Flow cytometry gating strategy is presented in supplemental Figure 2B. (D) Staining of peripheral blood smears day 3 postinfection showed the presence of aggregates (surrounded by dashed lines) in vehicle-treated mice but no aggregates (individual platelets indicated by arrow) in ketanserin-treated mice. Scale bar, 25 μm. (E-F) IFNR-KO mice were infected with DENV2 and treated with 8 mg/kg ketanserin or vehicle daily. (E) Ketanserin treatment rescued platelet counts during DENV infection. (F) Ketanserin treatment of DENV-infected mice resulted in a significant reduction in platelet activation compared with vehicle treated-controls at day 3 postinfection (n = 4-5 per group). (G) WT mice were infected with DENV2 and treated with ketanserin (8 mg/kg), sarpogrelate (3 mg/kg), or vehicle. Treatment with either 5HT2A antagonist rescued thrombocytopenia during DENV infection, compared with vehicle-treated controls. Data represent the mean and error bars represent the standard error of the mean. P values were determined by Student’s unpaired t test or by 1-way ANOVA (G). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Discussion
Thrombocytopenia is a classical presentation of dengue. Our data demonstrate that a rapid decrease in circulating platelets results from MC-derived serotonin acting on platelets, increasing their activation in response to DENV. Platelet activation results in aggregation and increased uptake in the spleen, leading to platelet destruction and thrombocytopenia in vivo in mice. Human platelets are also responsive to DENV-induced human MC-derived serotonin. Furthermore, targeting the MC-platelet axis using 5HT2 receptor antagonists reduces thrombocytopenia, highlighting a potential target for therapy.
Several mechanisms have been proposed for thrombocytopenia during dengue. Autoantibodies against platelets, potentially generated as a result of cross-reacting with DENV NS1, may cause thrombocytopenia.4 However, thrombocytopenia occurs not only during secondary infection but also during primary infection, when antibody responses are limited, supporting that there are other mechanisms. Thrombocytopenia is an early sign, occurring just after fever onset, suggesting that the virus itself or innate immune responses are involved. In humanized mice, infection of megakaryocytes led to production defects, although a caveat is that DENV induced a chronic rather than acute infection in humanized mice.46 Another ex vivo study suggested that DENV binding causes platelet apoptosis, leading to phagocytosis by macrophages.64 Here, we demonstrated that phagocytosis of activated platelets in the spleen is a critical component of DENV-induced thrombocytopenia in vivo. In patients with DENV, platelet counts also correlate with the levels of platelet activation, suggesting platelet activation contributes to thrombocytopenia.3,21,22 Our results indicate that host factors are crucial for potentiating the effect of DENV on platelets, as MCs are necessary for DENV to cause platelet activation in vivo. MC-produced serotonin was required for platelet activation and thrombocytopenia, as reconstitution of MC-deficient mice with MCs was sufficient to restore thrombocytopenia in DENV-infected animals. Reconstitution with MCs lacking serotonin synthesis did not recapitulate DENV-induced thrombocytopenia. Thus, MC-derived serotonin contributes significantly to platelet activation and thrombocytopenia during DENV infection.
Serotonin plays several roles in the periphery. Through its receptors on immune cells, serotonin can promote both pro-inflammatory and anti-inflammatory responses.65,66 Serotonin can also modulate metabolism by acting on adipose tissue.67,68 With regard to the activity of serotonin on platelets, it was noted that patients receiving selective serotonin reuptake inhibitors had platelet dysfunction.69,70 There are multiple activating receptors on platelets, including 5HT2A, and receptors for collagen and thrombin. Collagen and thrombin trigger activation through glycoprotein VI or protease-activated receptors, respectively.71,72 MC-derived tryptase could potentially influence platelets by cleaving protease-activated receptors, but it does not induce platelet activation.73 5HT2A can also potentiate platelet activation induced by other stimuli.54 In vitro DENV was shown to activate platelets through direct binding of virus to platelets.3 Consistent with this, we demonstrate that serotonin acts on platelets through 5HT2A, significantly enhancing their activation to DENV. However, neither serotonin nor DENV was sufficient, individually, to induce thrombocytopenia. In vivo, inhibitors that limit platelet activation and aggregation are produced, including prostacyclin.57,58 These are not present in cell culture, which may explain why we and others3,74 observe that DENV itself is sufficient to activate platelets in vitro. Activating platelets need to overcome an inhibitory threshold in vivo, which is important, as inappropriate activation could result in thrombosis and inflammation.75 Consistent with this, our results showed that serotonin overcame the inhibitory effects of a prostacyclin analog on DENV-induced platelet activation.
We have shown, in vitro and in vivo, that antagonists of the platelet 5HT2A serotonin receptor (ketanserin and sarpogrelate) effectively reduce serotonin-enhanced platelet aggregation and DENV-associated thrombocytopenia. Although the drugs tested can bind some other receptors with low affinity, both are highly selective for the 5HT2A receptor.76,77 For example, Ketanserin can also bind to 5HT2C, and sarpogrelate also binds to 5HT2B, yet both have higher specificity for 5HT2A, and neither 5HT2C nor 5HT2B is expressed on platelets,78 supporting that the activity of the drug on platelets is via the 5HT2A receptor. Because platelets can contribute to combating infections, we questioned whether 5HT-receptor antagonists could influence virus infection, but 5HT2A antagonism did not influence viremia. Because DENV infection is not increased by the drug treatment, it is unlikely to pose a risk of unsafely augmenting viremia. Conversely, our results support that reduced platelet activation is not merely an indirect result of reduced viral titers, as 5HT2A antagonism did not reduce DENV titers. Drugs targeting serotonin receptors are effective in reducing platelet activation in the context of vascular conditions such as ischemic stroke,43,44,79 and dengue represents a promising alternative use.
Although also frequently present during mild dengue, thrombocytopenia is a warning sign for progression to severe dengue. Platelet counts are lower and platelet activation is higher in patients with severe compared with mild dengue.21,22 Interestingly, MC activation is also enhanced during severe compared with mild dengue.28,29 During early infection, MCs are beneficial in clearing DENV, although the heightened activation of MCs during systemic infection, combined with their location surrounding blood vessels, can lead to vascular pathology.27-29 MCs are also recognized for their regulatory function at the neuroimmune juncture and are uniquely capable among immune cells of synthesizing and storing serotonin. Serotonin modulates platelet function,39 and products from cell lines with some attributes of MCs could activate platelets.31 MC products also promote platelet activation during other inflammatory diseases, such as in deep vein thrombosis.80 Although we observe that serotonin is specifically important for DENV-induced platelet activation, MCs make other products that influence hemostasis. Chymase, a MC-specific protease, can prolong bleeding time by affecting the coagulation cascade,81 but is not required for DENV-induced thrombocytopenia, as antagonism of serotonin receptors or specific ablation of MC-serotonin synthase are sufficient to block thrombocytopenia. MC-stabilizing drugs, which can also restrict thrombocytopenia during DENV infection, are commercially available, are used in the treatment of asthma, and are currently being tested for treatment of dengue vascular leakage. MC stabilizers prevent the exocytosis of granules and can also block platelet-independent mechanisms of DENV-induced vascular leakage.29 Our data also suggest that fluoxetine, used to deplete intracellular serotonin, can reduce platelet aggregation and activation during DENV, suggesting that a positive feedback loop exists downstream of 5HT receptors. However, pretreatment was required to deplete platelet serotonin, which has limitations as a therapeutic strategy. Because fluoxetine and other selective serotonin reuptake inhibitors are widely used antidepressive drugs, it raises the question of whether those taking it could be more resistant to DENV-induced thrombocytopenia. There are case reports describing patients taking fluoxetine before DENV infection, but its influence on the clinical course cannot be presumed, as they described an atypical disease course82 or discontinuation of the drug at the start of illness.83 Using inhibitors, we also have shown that serotonin enhances platelet activation downstream of 5HT receptors through serotonylation. As serotonylation occurs secondary to platelet activation, it is another potential target for therapy, although no drug is currently clinically available. One concern regarding the use of serotonergic drugs in dengue is that they could worsen bleeding by inhibiting platelet activation during thrombocytopenia. Current dengue treatment guidelines caution against the continued use of antiplatelet agents (such as aspirin) to reduce the bleeding risk.2 However, our data suggest that inhibiting thrombocytopenia during acute dengue could be beneficial, as mice did not experience increased vascular leakage during treatment with 5HT2A antagonists. Moreover, as the platelets of patients with dengue are activated and in an exhausted state,21 the potential for worsened bleeding with antiserotonergic drugs is low, although this would require additional testing.
Our findings highlight that both host and viral factors contribute to dengue thrombocytopenia. Importantly, targeting serotonin, a host factor, can ameliorate thrombocytopenia, revealing a potential therapeutic target of disease. Targeting of host factors has the added benefit of avoiding the potential of virus adaptation. Notably, other infections caused by hemorrhagic fever viruses, such as filoviruses and bunyaviruses, also induce thrombocytopenia84 and may be responsive to this therapeutic strategy. MCs are a previously unrecognized component of thrombocytopenia during dengue, via the release of serotonin, which promotes platelet activation through 5HT2A receptors and subsequent removal from circulation.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Michel Arock for providing the ROSA cell line, Axel Roers for providing the Mcpt5-cre mice, and Wilfried Saron for experimental advice and discussions. This work was funded by NMRC/CBRG/0084/2015 and start-up funding from Duke-National University of Singapore Medical School.
Authorship
Contribution: M.F.B.M. designed and performed the experiments, analyzed the data, and wrote the manuscript; C.K.M. designed and performed the experiments and analyzed the data; A.P.S.R. designed the experiments and wrote the manuscript; A.L.S.J. designed the experiments, analyzed the data, wrote the manuscript, and funded and supervised the project; and all authors approved the submitted version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for A.P.S.R. is Department of Pathology, Duke University Medical Center, Durham, NC.
Correspondence: Ashley L. St. John, Program in Emerging Infectious Diseases, Duke-National University of Singapore Medical School, 8 College Rd, Level 9, Singapore; e-mail: ashley.st.john@duke-nus.edu.sg.