

In the current issue of Blood, present the results of interesting studies confirming the utility of a novel approach to identify and validate potential targets of T-cell responses that could serve as potential therapeutic targets for cancer immunotherapy, including for hematologic malignancies.1
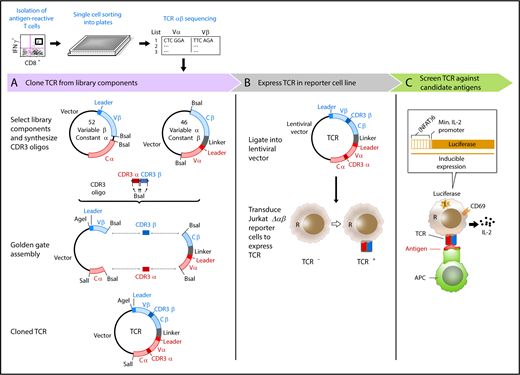
Overview of immunotherapy target discovery approach by Hu et al, including cloning, expression, and screening for T-cell activity against candidate antigens. APC, antigen-presenting cell; IFNγ, interferon-γ; IL-2, interleukin-2; L, lucerifase; Min., minimal; R, reporter. See Figure 1 in the article by Hu et al that begins on page 1911.
Overview of immunotherapy target discovery approach by Hu et al, including cloning, expression, and screening for T-cell activity against candidate antigens. APC, antigen-presenting cell; IFNγ, interferon-γ; IL-2, interleukin-2; L, lucerifase; Min., minimal; R, reporter. See Figure 1 in the article by Hu et al that begins on page 1911.
For millennia, humans have sought to divine the future, using approaches that range from dowsing for hidden water or predicting the course of events by diverse methods including tarot cards to astrology to palmistry. Lengthy tomes the length of modern medical textbooks have been written about many of these pseudoscientific approaches. Even in J. K. Rowling’s Potterverse, where conjuring, potion making, and magical herbology are considered standard and accepted disciplines, divination is considered an inexact science and is once described as “one of the most imprecise branches of magic.”2
Unfortunately, in our less magical world, the divination of putative targets of T-cell responses has similarly lagged behind its companion fields of vaccine development for pathogens, and the development of increasingly impressive immunotherapies using either checkpoint inhibitors or antigen-specific genetically modified T cells, including chimeric antigen receptor (CAR-T) therapies. The enthusiasm for the latter approach, in particular, is now palpable, with hematologists seeing truly unprecedented responses for patients receiving CD19-directed CAR-T therapies for both acute lymphoid leukemia in children and young adults and aggressive non-Hodgkin lymphoma (NHL) of older adults.3,4 In the coming year, it is likely that we will see additional approvals for CD19-targeted T-cell therapies, and very possibly an approved approach to target B-cell maturation antigen (BCMA) in myeloma patients,5 given promising early trial results.
Although these advances in hematology are truly electric, many of us are also concerned that the current buzz around CD19 and BCMA may fade if additional targets for these and other immunological therapies do not quickly follow. Our ability to further expand these approaches to a broad range of other targets for cancers is something we should not take for granted. Although a discussion of an ideal antigen is beyond the scope of this commentary, we can look at CD19 to know that it is an antigen widely expressed on healthy and malignant B cells. There is acceptable on-target toxicity (leading to transient B-cell aplasia and hypogammaglobulinemia) and significant, yet typically nonfatal, off-target effects that may include cytokine release syndromes and neurotoxicity.6 Although antigen escape may occur and is associated with relapse of both acute lymphoblastic leukemia and NHL, CD19 is typically expressed consistently enough to yield reliable responses in most treated subjects. Similarly, BCMA seems to have similar characteristics, making it a promising target in plasma cell malignancies. However, for immunotherapy to broadly succeed beyond a few selected blood cancers, the pace and quality of target antigen discovery must increase dramatically, or the limited availability of suitable targets will prove the greatest bottleneck to success, especially for solid tumors.
For this reason, the studies presented here by Hu and colleagues provide real hope that applying novel systems to identify and validate antigen discovery will yield a greater pipeline of targets for cancer-specific T cells and vaccines. The clever system devised by the investigators (see figure) includes 3 major steps: (1) using a functional measure of stimulated T-cell activity (eg, IFN-γ production), antigen-activated T cells were isolated and cloned using a novel and rapid single cell sequencing approach to identify the TCRαβ repertoire of reactive cells; (2) the newly identified TCRs were then expressed into a Jurkat line lacking a native TCR (following targeted CRISPR-Cas9 knockdown); and (3) the TCR expressing transduced cell line is then screened for reactivity against candidate antigens by reporter expression, validating their specificity.
In their elegant work, the authors first demonstrate that peripheral blood mononuclear cells from healthy donors could be stimulated with a broad commercial peptide mixture containing candidate antigens from multiple pathogenic viruses (“CEF”) and that this approach successfully identified and yielded demonstrably active virus-specific TCR sequences, including those recognizing cytomegalovirus (CMV), influenza, and Epstein-Barr virus. They then demonstrated that peripheral blood mononuclear cells from melanoma patients previously immunized with a personalized neoantigen vaccination could yield TCR sequences that were clearly recognizing relevant melanoma neoantigens. Finally, the authors turned to chronic lymphocytic leukemia (CLL) patients and showed that dominant TCRs could be identified that recognized candidate neoantigens from a putative CLL driver and confirmed its functional activity and HLA-restricted specificity.
It seems clear that this novel and systematic approach should yield relevant neoantigens that could result in vaccine and adoptive T-cell targets. However, there are caveats that should be noted. Our understanding of antigen recognition restricted by less commonly expressed HLA alleles lags behind our understanding of binding to common alleles, like HLA A2, which was first crystallized. Any expression system like this necessarily depends on characteristics of the reporter cells, which cannot exactly approximate the remarkable diversity of human T cells, which vary in their naive/memory differentiation, expression of costimulatory receptors that can attenuate their responses, and production of combinations of effector proteins, including cytokines. We have previously demonstrated that for CMV-specific T cells that the total number of cells (even that produce IFN-γ on cognate antigen stimulation) may poorly reflect a protective antipathogen response,7 and that single or combination functional signatures may better reflect viral reactivation risk.8 Despite these caveats, it is also very clear that the system devised here can be iteratively improved to better reflect native T-cell diversity and recognition, in order to yield increasingly better candidate TCRs most likely to have therapeutic value.
In summary, the novel and comprehensive approach outlined here has real potential to move the art of antigen discovery from its current place in the shadows of T-cell engineering to a more rapid and reliable approach to identify and validate targets for a broad range of human cancers. Given the current justified excitement about immunotherapy for blood cancers, there is a clear need for approaches like this one that can systematically identify novel targets for T-cell responses especially for solid tumors, including putative neoantigens. I am optimistic that studies like this one will help us fully unlock the promise of antigen-specific immunotherapy and move the previously inexact science of antigen discovery from the shadows into bright sunlight.
Conflict-of-interest disclosure: K.V.K. has served as an ad hoc scientific advisor to immunotherapy companies, including Kite/Gilead, Novartis, Juno/Celgene, and Atara Biotherapeutics.

