

Abstract
The traditional view of angiogenesis emphasizes proliferation and migration of vessel wall-associated endothelial cells. However, circulating endothelial progenitor cells have recently been shown to contribute to tumor angiogenesis. Here we quantify the relative contributions of endothelial and endothelial progenitor cells to angiogenesis using a mathematical model. The model predicts that during the early stages of tumor growth, endothelial progenitors have a significant impact on tumor growth and angiogenesis, mediated primarily by their localization in the tumor, not by their proliferation. The model also shows that, as the tumor grows, endothelial progenitors adhere preferentially near the tumor periphery, coincident with the location of highest vascular density, supporting their potential utility as vectors for targeted delivery of therapeutics. Model simulations of various antiangiogenic strategies show that those therapies that effectively target both endothelial and endothelial progenitor cells, either by restoring the balance between angiogenic stimulators and inhibitors or by targeting both types of cells directly, are most effective at delaying tumor growth. The combination of continuous low-dose chemotherapy and antiangiogenic therapy is predicted to have the most significant effect on therapeutic outcome. The model offers new insight into tumor angiogenesis with implications for the rational design of antiangiogenic therapy. (Blood. 2003;102:2555-2561)
Introduction
Angiogenesis plays a critical role in many physiologic and pathologic processes including tumor growth and metastasis.1 In the conventional view, angiogenesis is mediated by the local proliferation and migration of vessel wall-associated endothelial cells (ECs) in response to angiogenic growth factors.1 The genetic stability and accessibility to systemically delivered therapeutics of these ECs lining tumor blood vessels render them an attractive target for therapy.2 As a result, strategies targeting various stages of angiogenesis have been the subject of intense study.3
However, a growing body of experimental work suggests that circulating cells with the potential to differentiate into mature ECs,4 so-called circulating endothelial precursors or endothelial progenitor cells (EPCs), may also contribute to tumor angiogenesis.5-7 These recent studies suggest a newly emerging portrait of tumor angiogenesis—one in which new blood vessel growth arises from the combined contributions of both ECs and EPCs—and underscore the need for consideration of systemic as well as local events.
The hypothesis that both ECs and EPCs contribute to angiogenesis necessitates a new approach to quantify the roles of each and to characterize the interactions between them. The objective of this study was to quantify the contribution of EPCs through a mathematical analysis that synthesizes various aspects of this emerging paradigm of tumor angiogenesis into a single, coherent model. We show that such an undertaking is valuable for predicting the relative effectiveness of various antiangiogenic treatment strategies. The model also represents a flexible, modular framework useful for organizing and analyzing new quantitative data as it emerges, for testing hypotheses, and for guiding future experiments.
Methods
Here we briefly describe the governing equations underlying the mathematical model. The local and systemic contributions to angiogenesis (Figure 1) are described by a tumor growth and angiogenesis model and a physiologically based cell biodistribution model, respectively. The angiogenesis-related events occurring within the tumor comprise the local contribution (Figure 1). The systemic contribution consists of the angiogenesis-related events that occur throughout the circulatory system and in the bone marrow (BM; Figure 1). These 2 models are coupled by angiogenic growth factor concentration and circulating EPC population. (See the Supplemental Document link at the top of the article on the Blood website for a more detailed derivation of the equations as well as a description of the physical meaning underlying each of the parameters and the experimental data used to calculate all the parameter values.)

Model schematic illustrating the local and systemic contributions to angiogenesis. The human body is modeled as a series of organ compartments connected in an anatomic manner. The local contribution consists of the proliferation and migration of ECs (orange cells) in response to the local imbalance between angiogenesis stimulators and inhibitors. The angiogenesis stimulators produced by the tumor initiate the systemic pathway as well, which involves the mobilization of EPCs from the bone marrow. EPCs (blue cells) can localize in tumor vessels and contribute to angiogenesis.
Model schematic illustrating the local and systemic contributions to angiogenesis. The human body is modeled as a series of organ compartments connected in an anatomic manner. The local contribution consists of the proliferation and migration of ECs (orange cells) in response to the local imbalance between angiogenesis stimulators and inhibitors. The angiogenesis stimulators produced by the tumor initiate the systemic pathway as well, which involves the mobilization of EPCs from the bone marrow. EPCs (blue cells) can localize in tumor vessels and contribute to angiogenesis.
Tumor growth and angiogenesis model
In our model, the tumor initially consists of a small cluster of tumor cells and their pre-existing, co-opted microvascular network. As the tumor grows, the balance between angiogenic growth factors and endogenous antiangiogenic agents becomes disrupted, initiating angiogenesis.8 The differences in calculated production, degradation, and diffusion rates of angiogenic and antiangiogenic factors create zones of angiogenesis stimulation and suppression9 within the tumor. This imbalance between stimulators and inhibitors is represented by net angiogenic activity, α, defined such that angiogenesis is stimulated where α is greater than zero and suppressed elsewhere. By quantitatively comparing the predicted angiogenic activity with experimental perfusion data,10 we have formulated an empiric relationship between α and EC-derived vascular density:
In addition to their mitogenic activity, certain angiogenic factors may increase expression of adhesion molecules11 as well. Therefore we assume that for α greater than zero, adhesion of EPCs to the tumor vasculature is enhanced.4,12 The rate of accumulation of EPCs in the tumor is described by
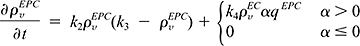
where the tumor growth rate, g, is dependent on the total vascular density
Physiologically based cell biodistribution model
In our model, angiogenic factors produced by the tumor travel to the BM where they stimulate the mobilization of EPCs.14,15 Once in the circulation, EPCs begin sampling the vascular beds of various organs, including the tumor. Following incorporation into the tumor microvasculature, EPCs enhance tumor growth (equation [Eq] 3) by contributing to new vessel formation.14 The biodistribution of angiogenic growth factors and circulating EPCs in the systemic circulation is described using a physiologically based biodistribution model.16 In this formulation, the concentration (Cji)of species conserved (j) in the vascular space of each organ (i) is described by
where the species conserved (j) is either angiogenic growth factors (α) or circulating EPCs (β). Blood flow rate (Qi) and vascular volume (Vv,i) are changing continuously in the tumor due to angiogenesis. The generation of angiogenic growth factors by the tumor (Gαi) depends upon the angiogenic activity (α), and EPCs are mobilized in the BM (Gβi) in proportion to the concentration of circulating angiogenic factors (Cαi).15 The concentration of EPCsCβi) is transformed into a source flux (qEPC) and passed to the tumor growth and angiogenesis model (Eq 2).
We focus on murine models since the bulk of available experimental data originates from mice. Murine models are also used in most preclinical in vivo experiments, facilitating direct comparison of model predictions with experimental data. For these reasons, our mathematical model simulates the tumor growth and early angiogenesis that ensues following the implantation of a small number of cancer cells into a mouse. Our model uses parameter values extracted or calculated from experimental studies (Supplemental Data, Section B). Consequently, most of the results shown represent a priori predictions of the model. The field of stem cell research is still in its infancy and, as such, there is a paucity of experimental data with which to make direct comparisons with model predictions. Thus, we perform quantitative comparisons whenever possible and point out qualitative agreements elsewhere.
Results
Role of endothelial progenitor cells in early tumor angiogenesis
As the tumor grows, the highest levels of angiogenic activity (α) occur near the periphery (Figure 2A) in qualitative agreement with experimental perfusion data10 (Figure 2A, inset). The dependence of vascular density on the imbalance between angiogenesis stimulators and inhibitors can be seen by comparing angiogenic activity (Figure 2A) with total vascular density (Figure 2B). The predicted total vascular densities agree with experimental values measured in tumors of comparable size17 (Figure 2C). As the tumor grows, a peak in the vascular density develops at the periphery, mirroring tissue perfusion profiles (Figure 2A, inset). The total vascular volume, determined by integrating the vascular density over tumor volume, also increases with tumor size (Figure 2B, inset) as expected.

EPC contribution to tumor angiogenesis.(A) The angiogenic activity α as a function of position for tumors of different radii: Rt ≈ 0.5 mm, t ≈ 10 days (d); Rt ≈ 1.0 mm, t ≈ 20 d; and Rt ≈ 1.5 mm, t ≈ 25 d. As the tumor grows, the highest angiogenic activity becomes localized near the periphery and extends beyond the tumor boundary for a distance approximately equal to the tumor radius, meaning that angiogenesis is stimulated in the adjacent normal tissue as well. Far from the tumor, α approaches zero, indicating a balance between angiogenesis stimulators and inhibitors resulting in a stable vascular network. The inset shows the correlation between experimental perfusion measurements10 and angiogenic activity. (B) Total vascular density as a function of position for tumors of different radii. The tumor initially has a radius of 300 μm and a vascular density equal to that of the normal tissue. Since the vascular density profiles are assumed to mirror the α profiles9 (A), the highest vascular density becomes localized near the periphery as the tumor grows. Vascular volume also increases with tumor size as expected (inset). (C) Model predictions of total vascular density agree well with experimental data17 during the early stages of tumor growth. Experimental data are given as means ± SD. (D) EPC-derived vascular density as a function of position for tumors of different radii. EPC proliferation makes only a modest contribution to vascular density compared with the case in which EPCs do not proliferate. The inset shows the predicted increase in EPC-derived vascular volume fraction as tumor size increases.
EPC contribution to tumor angiogenesis.(A) The angiogenic activity α as a function of position for tumors of different radii: Rt ≈ 0.5 mm, t ≈ 10 days (d); Rt ≈ 1.0 mm, t ≈ 20 d; and Rt ≈ 1.5 mm, t ≈ 25 d. As the tumor grows, the highest angiogenic activity becomes localized near the periphery and extends beyond the tumor boundary for a distance approximately equal to the tumor radius, meaning that angiogenesis is stimulated in the adjacent normal tissue as well. Far from the tumor, α approaches zero, indicating a balance between angiogenesis stimulators and inhibitors resulting in a stable vascular network. The inset shows the correlation between experimental perfusion measurements10 and angiogenic activity. (B) Total vascular density as a function of position for tumors of different radii. The tumor initially has a radius of 300 μm and a vascular density equal to that of the normal tissue. Since the vascular density profiles are assumed to mirror the α profiles9 (A), the highest vascular density becomes localized near the periphery as the tumor grows. Vascular volume also increases with tumor size as expected (inset). (C) Model predictions of total vascular density agree well with experimental data17 during the early stages of tumor growth. Experimental data are given as means ± SD. (D) EPC-derived vascular density as a function of position for tumors of different radii. EPC proliferation makes only a modest contribution to vascular density compared with the case in which EPCs do not proliferate. The inset shows the predicted increase in EPC-derived vascular volume fraction as tumor size increases.
As the tumor grows, EPCs localize near the periphery (Figure 2D), reflecting the greater opportunity for adhesion in this region due to increased angiogenic activity (Figure 2A) and higher vascular density (Figure 2B), and in agreement with the localization of BM-derived cells observed in experimental tumor models.18 The EPC-derived vascular density reveals that EPCs contribute significantly to tumor angiogenesis. During the initial stages of angiogenesis, the percentage of total vascular density that is EPC-derived (Figure 2D) increases from approximately 2% (Rt ≈ 0.5 mm) to 25% (Rt ≈ 1.0 mm), similar to the percentages of EPCs observed in granulation tissue19 and corneal neovascularization,20 respectively. As the tumor gets larger (Rt ≈ 1.5 mm), the fraction of EPC-derived vascular volume increases to approximately 40% in agreement with the number of EPCs identified in certain tumors.21 Proliferation of EPCs following incorporation into the tumor vasculature is predicted to exert only a modest effect on vascular density (Figure 2D). This implies that EPCs contribute to the early stages of tumor angiogenesis primarily through localization in the tumor rather than proliferation at the site of angiogenesis.
Role of endothelial progenitor cells in tumor growth
The model predicts that EPCs appreciably enhance tumor growth (Figure 3). When the EPC contribution is ablated and only vessel wall-associated ECs of the pre-existing vasculature are involved in angiogenesis, tumors exhibit a growth delay of 4.1 days (d) or 14%. The effect of EPCs becomes apparent only after 10 to 15 d of tumor growth, the delay associated with EPC mobilization from the BM and localization in the tumor. This agrees with studies of tumors grown in the Id1+/-Id3-/- mouse model in which EPC+ vessels were observed 14 d after tumor implantation.21

EPC contribution to tumor growth. Tumor growth curves for the case in which both ECs and EPCs contribute to angiogenesis and that in which the EPC contribution is neglected. Model predictions for the case in which both ECs and EPCs are present were generated by comparing with experimental tumor growth data10 for tumors of comparable size. When the EPC contribution is ablated, the tumor exhibits a growth delay of 4.1 d or 14%. (Growth delay is defined here as the difference in time required for 2 tumors growing at different rates to reach a given size, Rt ≈ 1.5 mm. The percentage indicates the fraction of the total time attributable to some intervention that retards growth.) Experimental data are given as means ± SD.
EPC contribution to tumor growth. Tumor growth curves for the case in which both ECs and EPCs contribute to angiogenesis and that in which the EPC contribution is neglected. Model predictions for the case in which both ECs and EPCs are present were generated by comparing with experimental tumor growth data10 for tumors of comparable size. When the EPC contribution is ablated, the tumor exhibits a growth delay of 4.1 d or 14%. (Growth delay is defined here as the difference in time required for 2 tumors growing at different rates to reach a given size, Rt ≈ 1.5 mm. The percentage indicates the fraction of the total time attributable to some intervention that retards growth.) Experimental data are given as means ± SD.
Interestingly, although the fraction of EPC-derived vasculature is initially small (Figure 2D, inset), it is sufficient to alter the rate of tumor growth (Figure 3). The increase in fraction of EPC-derived vascular density as the tumor gets larger (Figure 2D, inset) is reflected in a corresponding increase in tumor growth rate. For tumors larger than those simulated here, the balance between angiogenesis stimulators and inhibitors may shift and the primary tumor may begin to exert systemic suppression of angiogenesis,9 at which point EPC incorporation may follow a different pattern.
Effects of various antiangiogenic therapies on tumor growth
The primary cause of mortality and morbidity in cancer patients is metastasis.22 One potential application of conventional and antiangiogenic therapies is to delay the growth of metastases in patients with detectable primary tumors.2 Here we use the model to investigate the relative effectiveness of various antiangiogenic therapeutic strategies on small, clinically undetectable metastases. By varying the appropriate model parameters (Section B.3 in Supplemental Data), we have predicted the impact of therapeutic strategies that target various aspects of the local or systemic pathways (Figure 1; Table 1) on tumor growth.
We first consider antiangiogenic strategies that target primarily the local contribution to angiogenesis (Figure 1). One such approach involves targeting EC migration (Table 1). This encompasses therapeutics that inhibit matrix metalloproteinases (MMPs) or target integrins involved in angiogenesis-related migration, such as αvβ3 and αvβ5 (Eliceiri and Cheresh23 ). By inhibiting matrix degradation or cellular adhesion, these therapies prevent ECs in the tumor from migrating in response to growth factor stimulation. The model predicts that these approaches will result in a growth delay of 10.9 days (Figure 4A-B). In addition, the reduction in tumor vascular density relative to the untreated case is comparable with that observed in studies using αvβ3 blocking agents.24,25

Effect of various treatment strategies on tumor growth. (A) Effect of different antiangiogenic strategies on tumor growth. Those therapies that target both the local and systemic pathways, such as anti-VEGF Ab and continuous low-dose chemotherapy combined with anti-VEGFR-2 Ab, are most effective at delaying tumor growth. (B) Relative efficacy of the different antiangiogenic therapies as measured by growth delay. The model predicts a growth delay of 30% or 10.9 d for therapies targeting EC migration, 46% or 21.6 d for therapies targeting VEGF signaling, 55% or 31.2 d for antiangiogenic scheduling of chemotherapy, 61% or 38.7 d for those targeting VEGF, and 64% or 43.9 d for combined continuous low-dose chemotherapy and anti-VEGFR-2 Ab. Therapies on the left target primarily either the local or systemic contribution and increasingly target both contributions more effectively moving to the right. (C) Comparison of vascular density and angiogenic activity during anti-VEGF Ab therapy with the case of no therapy. Normalization of angiogenic activity (α → 0) decreases the vascular density closer to that in normal tissue and greatly delays tumor growth by targeting both the local and systemic pathways. The model predicts a marked reduction in tumor vascular volume during anti-VEGF Ab therapy compared with the case of no therapy (inset). (D) Effect of different chemotherapeutic dosing strategies on tumor growth. The maximum tolerated dose strategy exerts a primarily antitumor effect resulting in a growth delay of 4.9 d. Antiangiogenic scheduling of chemotherapy results in more effective tumor suppression with a growth delay of 55% or 31.2 d. The circulating levels of EPCs predicted during antiangiogenic scheduling of chemotherapy are markedly reduced relative to the case of no treatment (inset). For the case of antiangiogenic scheduling of chemotherapy, doses were administered every6das indicated by the arrows.
Effect of various treatment strategies on tumor growth. (A) Effect of different antiangiogenic strategies on tumor growth. Those therapies that target both the local and systemic pathways, such as anti-VEGF Ab and continuous low-dose chemotherapy combined with anti-VEGFR-2 Ab, are most effective at delaying tumor growth. (B) Relative efficacy of the different antiangiogenic therapies as measured by growth delay. The model predicts a growth delay of 30% or 10.9 d for therapies targeting EC migration, 46% or 21.6 d for therapies targeting VEGF signaling, 55% or 31.2 d for antiangiogenic scheduling of chemotherapy, 61% or 38.7 d for those targeting VEGF, and 64% or 43.9 d for combined continuous low-dose chemotherapy and anti-VEGFR-2 Ab. Therapies on the left target primarily either the local or systemic contribution and increasingly target both contributions more effectively moving to the right. (C) Comparison of vascular density and angiogenic activity during anti-VEGF Ab therapy with the case of no therapy. Normalization of angiogenic activity (α → 0) decreases the vascular density closer to that in normal tissue and greatly delays tumor growth by targeting both the local and systemic pathways. The model predicts a marked reduction in tumor vascular volume during anti-VEGF Ab therapy compared with the case of no therapy (inset). (D) Effect of different chemotherapeutic dosing strategies on tumor growth. The maximum tolerated dose strategy exerts a primarily antitumor effect resulting in a growth delay of 4.9 d. Antiangiogenic scheduling of chemotherapy results in more effective tumor suppression with a growth delay of 55% or 31.2 d. The circulating levels of EPCs predicted during antiangiogenic scheduling of chemotherapy are markedly reduced relative to the case of no treatment (inset). For the case of antiangiogenic scheduling of chemotherapy, doses were administered every6das indicated by the arrows.
We next consider strategies that target the local contribution to angiogenesis with secondary effects on the systemic mechanism. One such approach involves blocking angiogenic growth factor signaling, preventing endothelial cell proliferation and migration (Table 1). For the case of vascular endothelial growth factor (VEGF) in particular, this may be achieved by using an antibody (Ab) against VEGF receptor-2 (VEGFR-2) or small molecule receptor tyrosine kinase (TK) inhibitors. This approach targets the local stimulation of ECs primarily, but may interrupt the systemic mechanism as well by inhibiting the mobilization of EPCs, which have been characterized as VEGFR-2+ (Asahara et al4 and Hattori et al15 ). In addition, interfering with the proliferation and migration of ECs reduces the vascular surface area to which EPCs may adhere. These combined effects result in a growth delay of 21.6 d (Figure 4A-B) and a reduction in vascular density, in agreement with studies on SU5416 (Fong et al26 ).
A second group of strategies include those that target both the local and systemic mechanisms directly (Figure 1). One such approach involves blocking critical angiogenic growth factors, such as VEGF, using an anti-VEGF Ab (Table 1). Within the tumor, this restores the balance between angiogenesis stimulators and inhibitors reflected in a decrease in α (Figure 4C), which, in turn, decreases the vascular density (Figure 4C) and vascular volume (Figure 4C, inset). The local effect is mediated by a reduction in angiogenic activity that slows new vessel formation. The effect of this strategy on the systemic pathway is mediated by a decrease in the amount of circulating VEGF, reducing EPC mobilization from the BM. Model simulations predict a growth delay of 38.7 d (Figure 4A-B), showing that targeting both the local and systemic mechanisms proves substantially more effective than focusing on either one alone. Interestingly, our model predicts that strategies targeting integrins or VEGFR-2 are more effective initially, but that anti-VEGF Ab therapy ultimately results in a longer growth delay (Figure 4A).
Antitumor and antiangiogenic effects of chemotherapy on tumor growth
We consider 2 chemotherapeutic dosing strategies: maximum-tolerated dose and antiangiogenic scheduling (continuous low dose). The maximum-tolerated dose paradigm is expected to exert a primarily antitumor effect by targeting proliferating tumor cells. In contrast, periodic administration of lower doses of chemotherapy (antiangiogenic scheduling) is hypothesized to result in cytotoxicity among the proliferating fraction of ECs as well as the EPC subpopulation of BM-derived cells, thereby impacting both the local and systemic contributions to angiogenesis (Figure 1). The model predicts a modest growth delay of 4.9 d following administration of the maximum-tolerated dose compared with 31.2 d in response to antiangiogenic dosing (Figure 4D) as a result of the predicted circulating levels of viable EPCs (Figure 4D, inset). Our predicted tumor growth curves are qualitatively similar to experimental results obtained using larger tumors,27 supporting the hypothesis that the effect of antiangiogenic scheduling of chemotherapy may be mediated, at least in part, by its impact on the EPC contribution to angiogenesis.
The antiangiogenic effects of certain chemotherapeutic dosing strategies suggests that combining this approach with one of the other antiangiogenic strategies may prove effective at significantly delaying tumor growth (Table 1). An example of such a strategy is the concurrent administration of continuous low-dose chemotherapy to target the proliferating subpopulation of ECs and EPCs and anti-VEGFR-2 Ab to target the VEGF signaling pathway and abrogate the survival effects conferred by VEGF. The model predicts this combined treatment to be most effective of all those considered, leading to a growth delay of 43.9 d, extending the period of growth by 64% relative to untreated tumors and by 18% relative to continuous low-dose chemotherapy alone (Figures 4A-B,D), in agreement with experimental studies using this combination therapy.28
Discussion
Using physiologically definable parameters with values extracted from experimental studies, we have developed a mathematical model that captures the salient features of early tumor growth and angiogenesis. Our model provides the first quantitative description of an emerging portrait of angiogenesis, one that integrates both local and systemic influences, including the contribution of EPCs. The model predicts that the systemic mechanism makes a significant contribution to tumor angiogenesis and growth, in accord with recent studies.21
The maximum vascular densities are predicted near the tumor border (Figure 2B) and, consequently, EPCs localize preferentially near the tumor periphery (Figure 2D), underscoring their potential use as cellular vectors for delivering antiangiogenic or antivascular agents. Indeed, administration of BM-derived cells producing endostatin has been shown to result in significant tumor growth delay18 comparable with that predicted by the simulations of anti-VEGF Ab therapy (Figure 4A). This suggests that endostatin and anti-VEGF Ab therapy (Figure 4C) should have similar effects on α and supports the hypothesis that net angiogenic activity can be modified either by decreasing levels of angiogenesis stimulators or increasing levels of inhibitors.
The model predicts a relatively modest contribution of EPC proliferation to vascular density during the early stages of angiogenesis (Figure 2D), consistent with the results of in vitro proliferation assays. These assays reveal that, compared with ECs, subpopulations of BM-derived cells isolated and identified as EPCs have a much greater proliferative capacity29 but exhibit a delayed response to mitogenic stimulation. This delayed response is reflected in the predicted relative contribution of EPC proliferation to tumor growth. The bulk of the total vascular density arises from the proliferation of ECs and the localization of EPCs mobilized from the BM (Figure 2B,D). Despite the moderate contribution of EPC proliferation in the early stages of angiogenesis, our model predicts that EPCs still have an appreciable impact on tumor growth (Figure 3). Moreover, our predictions imply that a rational therapeutic approach aimed at delaying the growth or maintaining the dormancy of small tumors should target EPC mobilization in addition to proliferation—not proliferation alone.
The model simulations suggest that strategies that target both the local and systemic contributions, either by administering multiple drugs in combination or a single drug with an appropriate target, such as anti-VEGF Ab, are most effective at inhibiting tumor growth. However, although anti-VEGF Ab and continuous low-dose chemotherapy combined with anti-VEGFR-2 Ab both target the local and systemic pathways (Figure 1; Table 1), the model predicts that these 2 approaches lead to different therapeutic outcomes (Figure 4A-B). This difference owes to the distinct mechanisms underlying the effect of each approach on the systemic pathway. Continuous low-dose chemotherapy may be expected to target the proliferating fraction of the BM relatively indiscriminately. In contrast, anti-VEGF Ab should abrogate the mobilization of only those BM-derived cells that are VEGF responsive. Different model parameters correspond to each of these 2 mechanisms, leading to the predicted difference in therapeutic outcome.
Similarly, although both anti-VEGF Ab and anti-VEGFR-2 Ab therapy target VEGF signaling and affect, to some extent, both the local and systemic pathways (Figure 1; Table 1), these 2 approaches are predicted to result in remarkably different growth delays (Figure 4A-B), reflecting the underlying biologic effects of each. Anti-VEGFR-2 Ab inhibits signaling through VEGFR-2, which is responsible for stimulating EC proliferation and migration and may be involved in EPC mobilization. By sequestering soluble VEGF, anti-VEGF Ab inhibits signaling not only through VEGFR-2, but through VEGFR-1 as well. VEGFR-1 has been shown to mediate EC morphogenesis30 and increase production of urokinase plasminogen activator,31 which plays a role in degrading vascular basement membrane prior to EC migration. Furthermore, recent studies suggest that, in addition to VEGFR-2+ EPCs, VEGFR-1+ hematopoietic stem cells (HSCs) may also play a role in tumor angiogenesis.21 Activated VEGFR-1+ cells have been shown to produce angiogenic growth factors such as VEGF and platelet-derived growth factor (PDGF)32 that stimulate new vessel formation and enhance existing vessel stability. Hematopoietic cells may also support angiogenesis, at least during the early stages, through production of matrix metalloproteinases such as MMP-9 (Coussens et al33 ). As expected, recent experimental studies confirm that blocking both VEGFR-1 and VEGFR-2 is necessary to effectively inhibit the early stages of tumor growth and angiogenesis.21 The broader influence of anti-VEGF Ab compared with anti-VEGFR-2 Ab is captured by the model parameters and leads to the predicted difference in the impact of these 2 strategies on tumor growth. This example illustrates how 2 approaches to targeting the same signaling pathway may ultimately result in substantially different effects on tumor growth.
Antiangiogenic strategies that aim to restore the balance between angiogenesis stimulators and inhibitors, such as anti-VEGF Ab (Figure 4C), may have benefits beyond those predicted by the model. In addition to reducing vessel density, these approaches may reverse many of the pathophysiologic attributes of individual vessels, including aberrant morphologic features that hinder transport and functional abnormalities such as hyperpermeability. When administered optimally, anti-VEGF Ab therapy may improve the localization of drugs for which abnormal vasculature is a barrier to delivery and, by increasing tissue oxygenation, may enhance radiotherapy as well.34
In addition to VEGF, a number of other factors including angiopoietin-1 (Ang-1),15 placental growth factor (PlGF),35,36 and granulocyte-macrophage colony-stimulating factor (GM-CSF)37 also mobilize EPCs and HSCs from the BM and may undermine therapies that target VEGF alone. Redundancy in the production of angiogenic factors is an important consideration in the design of therapies targeting vessel wall-associated ECs as well. A more comprehensive therapy may involve combining continuous low-dose chemotherapy with a receptor TK inhibitor that targets basic fibroblast growth factor (bFGF) and PDGF signaling in addition to VEGF signaling,38 instead of anti-VEGFR-2 Ab, which targets VEGF signaling alone. Our analysis underscores the importance of understanding which growth factors are produced by a given tumor when prescribing treatment and illustrates the usefulness of our model for aiding in the rational design of future antiangiogenic therapeutic strategies.
Importantly, the effect of angiogenesis inhibitors on EPCs has been investigated for only a few agents.39 The significance of the EPC contribution to vascular density predicted by our model emphasizes the importance of exploring the impact of various antiangiogenic agents on EPCs in order to improve the design of therapeutic strategies. For example, antiangiogenic therapy targeting migration may affect EPCs as well as ECs. But whether this effect would be exerted at the site of angiogenesis, in the BM, or both is not known. A novel approach to targeting the EPC contribution to angiogenesis would be to assess the integrin expression profile of EPCs and design therapeutics to target those that mediate adhesion. This strategy could be administered in combination with therapy targeting the integrins important for mediating the migration of ECs, such as αvβ3 or αvβ5 (Eliceiri and Cheresh23 ), thereby debilitating both the local and systemic contributions to tumor angiogenesis.
A substantial obstacle plaguing the search for clinically effective antiangiogenic therapies has been the difficulty involved in assessing their activity in vivo and determining a relevant biologic end point. EPCs may serve as useful surrogate markers of angiogenesis,40 but in order to exhaust this potential, a quantitative framework is needed to analyze and interpret experimental data in the context of antiangiogenic therapy. Our model serves as a tool for this purpose. Specifically, we are able to assess the extent to which a given therapeutic strategy would be expected to impact circulating levels of EPCs and the subsequent effect on tumor growth and angiogenesis.
The present picture of angiogenesis is complex, involving numerous stimulators and inhibitors as well as several cell types, and will likely become even more complicated as the intricacies of the signaling pathways underlying the effects of the various growth factors, inhibitors, and their receptors begin to surface. These elements can easily be embedded in the framework presented here as they become known to yield a model capable of resolving the angiogenic process in finer detail and potentially exposing new, more effective targets for therapy. For example, future versions of the model can incorporate the ability of other growth factors such as Ang-1 (Hattori et al15 ) to mobilize EPCs from the BM and quantify the potential contribution of the production of chemokines and cytokines by BM-derived cells.41 In addition, the physiologically based compartmental approach used in this study represents a flexible and versatile framework that may be adapted to a wide variety of applications beyond those considered here. These include studying the role of EPCs in other physiologic or pathophysiologic conditions involving angiogenesis1 and exploring the contribution of other adult stem or progenitor cells to organogenesis,42 regenerative medicine,43 and tissue engineering.44,45
Prepublished online as Blood First Edition Paper, May 29, 2003; DOI 10.1182/blood-2003-02-0365.
Supported by a Bioengineering Research Partnership grant (R24 CA85140) and a Program Project grant (P01 CA80124) to R.K.J.
B.R.S. and C.M. contributed equally to this work.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
The authors wish to thank J. Baish, E. di Tomaso, D. Duda, B. Fenton, C. Mouta Carreira, S. Ramanujan, T. Roose, H. D. Suit, and L. Xu for helpful discussions.

