

Abstract
Alloimmunization against red blood cell antigens and delayed hemolytic transfusion reaction (DHTR) are major barriers to transfusion in sickle cell disease (SCD). In SCD, DHTR is a potentially life-threatening. Blood group polymorphism in SCD patients, who are of African ancestry and frequently exposed to antigens they do not carry; an inflammatory clinical state; and occasional transfusion in acute situations are risk factors for alloimmunization and DHTR. In patients at risk, the transfusion indication must be balanced against the risk of developing DHTR. However, when transfusion is absolutely necessary, protocols combining the prevention of exposure to immunogenic antigens with immunosuppressive treatments must be implemented, and patients should be carefully monitored during posttransfusion follow-up. This close monitoring makes it possible to diagnose hyperhemolysis as soon as possible; to avoid retransfusion, which can exacerbate hemolysis; and to administer specific treatments, such as anticomplement therapy, in severe cases. Finally, in patients with severe disease, hematopoietic stem cell transplantation may be indicated. However, transfusion is also required in this context, and its management is complex because these risks must be taken into account.
We discuss these issues here, based on a clinical case. With the exception of consensual prophylactic red cell antigen matching for ABO/RhD and Rh(C, E or C/c, E/e), the recommendation of these measures is conditional because certainty concerning the evidence of an effect is very low. This lack of evidence highlights the need for improving our understanding of transfusion complications in SCD and developing preventive strategies that take into account the mechanisms underlying hyperhemolysis syndrome.
Learning Objectives
To know the risk factors of red blood cell alloimmunization and delayed DHTR in SCD
To describe the options for managing complex transfusion situations in an SCD patient when transfusion and/or hematopoietic stem cell transplantation are absolutely indicated
Introduction
Sickle cell disease (SCD) is the most common inherited red blood cell (RBC) disorder in individuals of African descent. Transfusion remains a major treatment, though with a certain risk in SCD. Delayed hemolytic transfusion reaction (DHTR) occurs in about 4% of adult patients receiving occasional transfusions.1 Delayed hemolytic transfusion reaction is an umbrella term encompassing posttransfusion hemolysis of all degrees of severity, with the potentially fatal development of hyperhemolysis occurring in the most severe cases.2,3 Indeed, 5% of the 266 deaths analyzed in SCD adults were due to DHTR.4
The major cause of DHTR is RBC alloimmunization because of differences in blood groups between donors of Caucasian origin and recipients of African ancestry. Another risk factor is the inflammation status at the time of transfusion during acute complications.3 However, many cases with no detectable antibodies are described, calling into question the underlying mechanism.5-10
In the most severe cases, the patient develops hyperhemolysis, with lower levels of hemoglobin (Hb) after transfusion than before, due to the destruction of transfused and autologous RBCs and, in some cases, profound reticulopenia.11 The severity of the condition is due to major intravascular hemolysis with the physiological protection against free Hb (haptoglobin) and free heme (hemopexin, heme oxygenase) being overwhelmed. Accumulation of free heme generates oxidative stress, triggering symptoms of a vaso-occlusive crisis (VOC) and accentuating the damage to vascular endothelial cells. Whether occurring through the classical or alternate pathways, complement activation, which results in membrane attack complex formation, is the predominant mechanism of hyperhemolysis. The patient's RBCs are destroyed because they are sensitive not only to oxidative stress but also to the nonspecific binding of activated complement fractions.12,13
Prevention of this accident is currently based on the prevention of alloimmunization but also involves immunosuppressive therapy. The treatment of this syndrome, particularly in severe cases, may require drugs acting on the complement activation cascade, such as eculizumab.14-17
We present here the clinical case of a patient with history of hyperhemolysis with antibody development for whom transfusion support continued to be indicated because of a severe symptomatic disease that could not be treated with hydroxycarbamide. The decision was made to perform hematopoietic stem cell transplantation (HSCT), which caused additional transfusion challenges.
THE CLINICAL CASE
A 15-year-old boy was referred to our department for highly symptomatic SCD refractory to hydroxycarbamide. In the last 2 years, he experienced 5 acute chest syndrome (ACS) and was hospitalized monthly for VOC. His RBC phenotype was B, D+ C-E- c+ e+, K-, Fya-Fyb-, Jka-Jkb+, M-N+ S- s. During a systematic antibody screening test without evidence of hemolysis, anti-Jka antibodies were detected after 3 transfusion episodes with prophylactic Rh- and K-matched units: one before cholecystectomy (3 RBC units) and two for treatment of ACS (2 RBC units each). After development of anti-Jka antibodies, a fourth transfusion to treat ACS was performed with 2 C-E-, K- and Jka- RBC units, with additional prophylactic matching for FY (Fya-Fyb-) and Ss (S-) leading to an immediate posttransfusion Hb level of 10.4 g/dL and 30% HbA. Ten days later, the patient developed joint pain, fever, jaundice, and hemoglobinuria. His Hb level fell to 6.4 g/dL (pretransfusion level: 8 g/dL), bilirubin rose from 18 (pretransfusion level) to 115 mmol/L and LDH level rose from 534 (pretransfusion level) to 1156 IU/L. The symptoms mimicked VOC, but the presence of hemoglobinuria and the clearance of HbA (15%) associated with the rapid rise in percentage of sickle cell Hb (HbS) led to a suspicion of hyperhemolysis. The patient received only supportive care (hydration, oxygenation, analgesic opioids), and the symptoms resolved. During the course of DHTR, antibody screening test revealed only the already known anti-Jka antibodies, but anti-M antibodies were detected 1 month later.
Given the severity of the disease, a transfusion program to prevent VOC recurrences18,19 followed by haploidentical HSCT was considered during a national multidisciplinary pediatric care meeting. A decision was made on a transfusion program: C-E-, K-, Fya-Fyb-, Jka-, S- and M- units along with prophylactic immunosuppression. Rituximab was administered (375 mg/m2 via IV) 10 days before the first transfusion. During the transfusion program, additional rituximab was administered to achieve continuous B-lymphocyte depletion. No adverse transfusion reaction occurred during the 6-month program.
The phenotype of the graft donor (the patient's mother) was O, Jka+, M+. The risk of an acute hemolytic reaction triggered by the donor's anti-B antibodies at the time of stem cell infusion was prevented by plasma reduction of the bone marrow graft. Recipient anti-Jka and anti-M antibodies were no longer detectable by the time of HSCT, but RBC reduction on the bone marrow graft was recommended to prevent restimulation by the donor's RBCs. HSCT was performed after a reduced-intensity conditioning regimen. During the preconditioning phase and after HSCT, the patient was transfused with O, C-E-, K-, Jka-, Fya-, S- and M- RBC units. The immunohematological work up remained negative. However, the patient experienced pure red-cell aplasia not of immunohematological origin and was dependent on RBC transfusion for 6 months after transplantation. He is now well and displays full engraftment with no acute or chronic graft versus host disease.
How to prevent posttransfusion hemolysis in patients with SCD
In our case, the patient initially developed antibodies after transfusion of 7 RBC units matched for Rh and K, reflecting his high- responder status. He presented only a delayed serologic reaction, as the development of the anti-Jka antibody was not associated with posttransfusion hemolysis. However, the patient was at risk of developing new antibodies and DHTR. The DHTR risk has been shown to increase with various factors: transfusion for an acute indication, history of immunization, history of DHTR, and transfusion with a smaller cumulative number of transfused units.1,5 In the case described, the patient had all the known risk factors except prior DHTR. In accordance with current transfusion recommendations,20,21 along with a Jka-negative protocol to take into account the preformed antibody, matching was extended to Fya and S antigens to prevent formation of new antibodies. In this case, the extended phenotype of the patient was known and previously performed with serologic techniques. As recommended, the patient had also benefited from a molecular work up to detect partial antigens and rare blood in the Rh blood group, which should also have been taken into account earlier. However, no variant was found. Despite this prophylaxis, the patient developed posttransfusion hemolysis with the appearance of anti-M antibodies. M-antigen matching is not included in the prevention of immunization at our center unless anti-M antibodies are already detectable or known in history. However, anti-M antibodies may be involved in DHTR, as shown by the most recent series of pediatric DHTR cases.9,10 Then, considering the M antigen in already immunized patients can be an issue. Rituximab, which can be used to prevent alloimmunization in patients with history of severe DHTR, might have prevented the production of this new antibody.22 This case highlights the complexity of decision-making in such cases and demonstrates that the risk of antibody development and the clinical significance of the antibodies associated with hyperhemolysis remain poorly understood.23 Indeed, in the context of DHTR, when antibodies are detected, they may be of any type and have no correlation with severity.7
How to manage patients with history of hyperhemolysis
The complex patients in France are discussed at a multidisciplinary consulting meeting with a panel of clinicians and transfusion pathologists specializing in SCD (Figure 1). The first aspect discussed is always the risk/benefit ratio of continuing transfusion, particularly for classical indications, such as the preoperative prevention of SCD symptoms. In the case described here, the clinical situation was reevaluated, and the continuation of programmed transfusions was considered the only appropriate treatment for this symptomatic patient before HSCT.
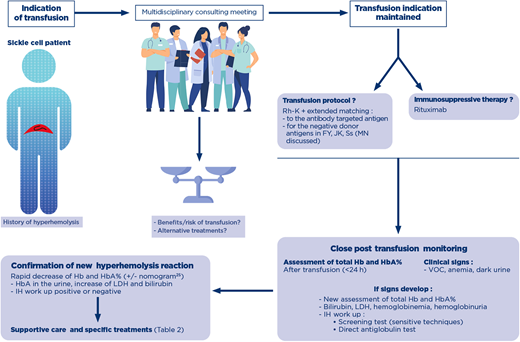
Decision making process for the indication of transfusion in a patient with history of hyperhemolysis. A patient with history of posttransfusion hyperhemolysis is at risk for recurrence of the syndrome, with or without detectable antibodies. When a transfusion is indicated, a shared decision-making process assesses the risk/benefit of transfusion, and if maintained, establishes the transfusion protocols in terms of RBC antigen matching and the use of immunosuppressive therapy. Close monitoring of the posttransfusion phase is absolutely necessary, and assessment of total Hb and HbA% immediately after transfusion will help diagnose posttransfusion hyperhemolysis with new assessment of these parameters. If clinical signs appear, evaluation of hemolytic biological parameters and an IH workup are performed. A negative IH workup does not rule out the diagnosis. After confirmation of hyperhemolysis, supportive care and specific treatments are considered. Hb, hemoglobin; IH, immunohematology; LDH, lactate dehydrogenase; RBC, red blood cell; VOC, vaso-occlusive crisis.
Decision making process for the indication of transfusion in a patient with history of hyperhemolysis. A patient with history of posttransfusion hyperhemolysis is at risk for recurrence of the syndrome, with or without detectable antibodies. When a transfusion is indicated, a shared decision-making process assesses the risk/benefit of transfusion, and if maintained, establishes the transfusion protocols in terms of RBC antigen matching and the use of immunosuppressive therapy. Close monitoring of the posttransfusion phase is absolutely necessary, and assessment of total Hb and HbA% immediately after transfusion will help diagnose posttransfusion hyperhemolysis with new assessment of these parameters. If clinical signs appear, evaluation of hemolytic biological parameters and an IH workup are performed. A negative IH workup does not rule out the diagnosis. After confirmation of hyperhemolysis, supportive care and specific treatments are considered. Hb, hemoglobin; IH, immunohematology; LDH, lactate dehydrogenase; RBC, red blood cell; VOC, vaso-occlusive crisis.
In a patient with a history of hyperhemolysis, when new transfusions cannot be avoided because of a severe disease, extended matching for RBCs for FY, JK and Ss is always indicated, and rituximab should always be provided, especially if the patient has developed hyperhemolysis with detectable antibodies, in accordance with a conditional recommendation of the ASH guidelines.20 (Table 1). There is currently no other widely used preventive measure against alloimmunization and DHTR. A retrospective study suggests that rituximab can be safely used for preventing DHTR in patients with a previous history of DHTR and detected antibodies. However, despite rituximab prophylaxis, DHTR recurred in about 20% of the patients, albeit at moderate severity and without detectable antibodies, confirming the multifactorial origin of this syndrome.23
The guidelines of the American Society of Hematology (ASH) discussed in this review
| Recommendations or suggestions of ASH | Grading of recommendations | Remarks | |
|---|---|---|---|
| Questions 3 and 4: Use of immunosuppressive therapy20 | ASH SUGGESTS: Immunosuppressive therapy over no immunosuppressive therapy for high risk of acute hemolytic transfusion reaction or severe history of DHTR | Conditional recommendation based on very low certainty in the evidence about effects | Share decision-making process is critical to weigh the potential benefits and harms associated with transfusion versus the effect of ongoing SC symptoms |
| ASH SUGGESTS: Immunosupressive therapy over no immunosuppressive therapy in patients with DHTR and ongoing hyperhemolysis | Conditional recommendation based on very low certainty in the evidence about effects | Share decision-making process Immunotherapy should be initiated promptly if ongoing hyperhemolysis with: - First line: IVIG and high dose steroids - Second line: Eculizumab - Rituximab only to prevent additional antibodies Precipitation of VOC with steroids should be considered |
| Recommendations or suggestions of ASH | Grading of recommendations | Remarks | |
|---|---|---|---|
| Questions 3 and 4: Use of immunosuppressive therapy20 | ASH SUGGESTS: Immunosuppressive therapy over no immunosuppressive therapy for high risk of acute hemolytic transfusion reaction or severe history of DHTR | Conditional recommendation based on very low certainty in the evidence about effects | Share decision-making process is critical to weigh the potential benefits and harms associated with transfusion versus the effect of ongoing SC symptoms |
| ASH SUGGESTS: Immunosupressive therapy over no immunosuppressive therapy in patients with DHTR and ongoing hyperhemolysis | Conditional recommendation based on very low certainty in the evidence about effects | Share decision-making process Immunotherapy should be initiated promptly if ongoing hyperhemolysis with: - First line: IVIG and high dose steroids - Second line: Eculizumab - Rituximab only to prevent additional antibodies Precipitation of VOC with steroids should be considered |
DHTR, delayed hemolytic transfusion reaction; IVIG, intra veinous immunoglobulin; SC, sickle cell; VOC, vaso-occlusive crisis.
All patients with a history of hyperhemolysis undergoing transfusion again should be monitored closely to ensure that any new episode is detected as soon as possible so that ad hoc treatments can be rapidly initiated at specialist facilities. (Figure 1).
The diagnosis of posttransfusion hemolysis
Recurrence or appearance of VOC shortly after transfusion, dark urine, onset or worsening of anemia, or increase in LDH concentration should alert professionals to the likelihood of DHTR. This first step—recognition—is crucial to prevent further transfusions, which would exacerbate hemolysis, and for the initiation of treatment before irreversible multiple organ failure develops. Diagnosis is not dependent on immediate evidence of newly formed antibodies. In our case, the anti-M antibody was detected in the plasma 30 days after the trigger transfusion. However, no antibodies are ever detected in many cases, even some time after the DHTR. The HbA decrease relative to the values obtained immediately after transfusion is a key parameter for the confirmation of posttransfusion hemolysis. A nomogram can facilitate diagnosis if assessments of total Hb and HbA% immediately after transfusion are available.24 In a study on children performed at 1 institution, HbA clearance was also calculated based exclusively on the volume of RBCs transfused and the hematocrit of the units.25 The appearance of HbA in the urine with a worsening of anemia may also indicate ongoing DHTR. Finally, a rapid decrease of 25% or more in total Hb levels with respect to pretransfusion levels should raise suspicions of DHTR. The diagnosis of DHTR and the elimination of other causes of acute anemia, such as splenic sequestration, autoimmune hemolytic anemia, acute hemolytic sickle cell crisis, and extensive bone marrow necrosis, are crucial because a history of DHTR affects treatment decisions, with patients subsequently receiving the smallest possible number of transfusions.
How to treat a patient with hyperhemolysis (Table 2)
In this case, the patient received only supportive care and did not require additional transfusions. In a recent pediatric series, 15% of patients received no treatment, whereas the others received EPO, rituximab, and/or eculizumab. Corticosteroids were also administered, but only for patients undergoing new transfusions due to life-threatening anemia.9 In another series of 37 pediatric cases, 11 patients received supportive care only; the other patients received immunosuppressive therapy. Additional transfusion was required for 17 patients. In this report, treatment with a high dose (2 mg/kg) of corticosteroid in addition to transfusion made it possible to maintain Hb levels after transfusion.10 Finally, in a series of cases in young adults,26 66% of patients received corticosteroids. By contrast, corticosteroid treatment is systematically avoided in the DHTR setting at some adult facilities due to the risk of hyperviscosity and new VOC.27 However, in most of the retrospective case studies, the patients received different treatments simultaneously, making it difficult to determine which of the them was actually effective.28
Current and potential treatments of posttransfusion hyperhemolysis based on putative mechanisms
| Goals of treatments | Treatments currently used | Potentially useful drugs |
|---|---|---|
| Supportive care | ||
| Hydratation, analgesic, oxygenation | ||
| Stimulation of erythropoiesis | ||
| Erythropoietin | ||
| Iron | ||
| Folates | ||
| Vitamin B12 | ||
| Inhibition of RBC destruction | ||
| Macrophages/antibody-mediated | ||
| Intravenous immunoglobulin | ||
| Complement cascade activation | ||
| Eculizumab | ||
| Other anti-C: C1 inhibitors39 | ||
| Elimination of toxic molecules from the plasma | ||
| Heme | Plasmapheresis | |
| Hemopexin40 | ||
| Hemoglobin | Plasmapheresis | |
| Haptoglobin40 | ||
| Antibodies | Plasmapheresis | |
| IgG endopeptidase41 | ||
| Activated fractions of complement, cytokines, other involved molecules | Plasmapheresis | |
| Anti-inflammatory action | ||
| Corticosteroids (balanced with vaso- occlusive risk) | ||
| Tocilizumab (case reports) | ||
| Safety of additional transfusion | ||
| Transfusion + rituximab | ||
| Transfusion + corticosteroids (balanced with vaso-occlusive risk) | ||
| Transfusion + Daratumumab (case report)42 | ||
| Transfusion + Bortezomib | ||
| Various goals | ||
| Bortezomib + Hemopure (case report)43 |
| Goals of treatments | Treatments currently used | Potentially useful drugs |
|---|---|---|
| Supportive care | ||
| Hydratation, analgesic, oxygenation | ||
| Stimulation of erythropoiesis | ||
| Erythropoietin | ||
| Iron | ||
| Folates | ||
| Vitamin B12 | ||
| Inhibition of RBC destruction | ||
| Macrophages/antibody-mediated | ||
| Intravenous immunoglobulin | ||
| Complement cascade activation | ||
| Eculizumab | ||
| Other anti-C: C1 inhibitors39 | ||
| Elimination of toxic molecules from the plasma | ||
| Heme | Plasmapheresis | |
| Hemopexin40 | ||
| Hemoglobin | Plasmapheresis | |
| Haptoglobin40 | ||
| Antibodies | Plasmapheresis | |
| IgG endopeptidase41 | ||
| Activated fractions of complement, cytokines, other involved molecules | Plasmapheresis | |
| Anti-inflammatory action | ||
| Corticosteroids (balanced with vaso- occlusive risk) | ||
| Tocilizumab (case reports) | ||
| Safety of additional transfusion | ||
| Transfusion + rituximab | ||
| Transfusion + corticosteroids (balanced with vaso-occlusive risk) | ||
| Transfusion + Daratumumab (case report)42 | ||
| Transfusion + Bortezomib | ||
| Various goals | ||
| Bortezomib + Hemopure (case report)43 |
There is still little consensus about DHTR management. It differs between adults and children and between the different centers treating patients; however, DHTR management also differs based on the treatments available, as some, such as eculizumab, are expensive and difficult to obtain at some facilities.
Aside from supportive care, current treatments are based on the putative mechanisms underlying DHTR and the consequences of the release of RBC content. RBC production has to be stimulated by EPO and the different RBC destruction pathways inhibited. IVIG treatment is considered against the macrophage- mediated destruction of RBCs induced by antibodies, and anticomplement treatment is considered against the complement activation through the classical and alternative pathways. Eculizumab is the only anticomplement agent used to date in the context of DHTR.29 However, it must be administered at the very start of hyperhemolysis to prevent irreversible multiple organ failure. Another goal of treatment is to eliminate free heme and free hemoglobin released in the plasma, which can have deleterious effects on the endothelial cells in the vessels. Plasma exchange is a good option; however, extracorporeal volume must also be considered, as it may cause a further decrease in hemoglobin level.30 Other drugs for removing free heme from the plasma, such as hemopexin, could be considered in the future. Other drugs have been used to treat hyperhemolysis, such as Tocilizumab, an anti–IL6 receptor agent that lowers the levels of inflammatory markers thought to be involved in the pathophysiology of severe DHTR.3132
Finally, in some cases of profound anemia and organ hypoxia, further transfusions are inevitable. If transfusion is indicated, extended matching (Fy, Jk, Ss) is recommended in addition to rituximab prophylaxis, even if there are no detectable antibodies. Pediatric clinicians also consider administering a short course of corticosteroids to patients undergoing transfusion in this context because of this treatment's anti-inflammatory and immunosuppressive effects.910
The challenges of transfusion in the context of hematopoietic stem cell transplantation
Hematopoietic stem cell transplantation (HSCT), as in the case described, is the only curative treatment in patients with severe SCD-related complications, especially when transfusion difficulties are encountered due to the presence of multiple antibodies, a rare blood type, or a history of severe DHTR.33 In SCD patients undergoing HSCT, transfusion support is required before the initiation of the conditioning regimen, with RBC exchange transfusion to decrease HbS% to about 30% to prevent vaso-occlusive complications and also to sustain the postconditioning aplasic phase. In nonmyeloablative HSCT transplantation, which is frequently sufficient to cure the disease, stable mixed chimerism is associated with a risk of immunohematological complications.34 The coexistence of donor and recipient immune cells results in a risk of RBC antibody production by the immune cells of both donor and patient. In a series of 61 patients, 3 patients developed antibodies against the donor or recipient RBCs after HSCT. The complications observed ranged from nonsevere adverse effects to near-fatal hemolysis.35 During HSCT for SCD, in addition to the usual management of ABO-mismatches, clinicians must also consider complicated transfusion situations, such as the production of multiple alloantibodies by the patient, rare blood groups, and the prevention of DHTR.36 Thus, once the indication for HSCT has been validated, and the donor has been selected, the feasibility of transfusion support should be evaluated at 2 levels. First, the availability of extended phenotype-matched RBC or rare blood, compatible with both donor and recipient, should be checked. HSCT patients with a history of RBC alloantibody have been shown to receive significantly more RBC units during HSCT than non-alloimmunized patients.37 Secondly, prevention of DHTR by immunosuppressive therapy in patients with a history of DHTR should be discussed. In a retrospective series of 34 adolescents and young adults, 15% required prophylaxis for DHTR during preconditioning transfusion support.34 With the increase in indications for HSCT in young and older patients,38 we are likely to see a parallel increase in the frequency of complex transfusion situations.
Conclusion
In SCD, transfusion can induce reactions ranging from a delayed serologic reaction sometime after transfusion, with no evidence of hemolysis, to the development of posttransfusion hemolysis, with the most severe cases presenting a hyperhemolysis syndrome. The prevention of these reactions is currently based solely on avoiding exposure to immunogenic antigens, based on blood group stratification compatibility according to the patient's history, and the administration of immunosuppressive treatments.
However, even with such prophylaxis, some cases of DHTR occur and their pathophysiology remains unclear, as no RBC antibodies are detected in many cases.
The posttransfusion monitoring of patients with history of alloimmunization and/or posttransfusion hemolysis is highly important to ensure that specific treatment can be initiated rapidly if signs of hemolysis develop. Finally, when HSCT is indicated, transfusion protocols and the work-up on the bone marrow graft should take into account both the RBC phenotype of the graft donor and the complex transfusion situation of the patient. However, most of the measures described in this review are qualified by the American Society of Hematology (ASH) guidelines as conditional recommendations based on the very low certainty of the evidence for an effect.19 (Table 1) This lack of evidence highlights the importance of improving our understanding of transfusion complications in SCD and of developing suitable methods for preventing hyperhemolysis based on the underlying mechanisms.
Acknowledgments
We thank Nathalie Dhedin, MD, APHP, Saint Louis Hospital, Paris, France, and Anne-Claire Leprêtre, MD, Etablissement Français du Sang, Saint Louis Hospital, Paris, France, for the joint reflections on the HSCT topic covered in this manuscript. We thank Emilie da Silva Rosa, Etablissement Français du sang, Ivry, France, for the graphic design of the figures.
Conflict-of-interest disclosure
France Pirenne: No conflict related to this review
Corinne Pondarré: honoraria for Novartis and expert consultancy for Addmedica
Off-label drug use
France Pirenne: Nothing to disclose.
Corinne Pondarré: Nothing to disclose.
