

Abstract
Thrombotic complications are the primary contributor to morbidity and mortality in essential thrombocythemia (ET) and polycythemia vera (PV). Cytoreductive therapy is the main tool for primary or tertiary thrombosis prevention in these diseases. In general, high-thrombotic-risk patients and those with symptoms that may be ameliorated from cytoreductive therapy are candidates for this treatment, although the decision is highly individualized. Approved options for cytoreduction in ET and PV include hydroxyurea, long-acting interferons, anagrelide in ET, and ruxolitinib in PV. Selecting the ideal agent requires careful consideration of the toxicity profiles and individual treatment goals. In this review the existing literature on cytoreductive decisions in ET and PV is summarized, with an emphasis on risk-stratification, highlighting the need for personalized care in order to maximize the benefit of these therapies while minimizing toxicities.
Learning Objectives
Understand the current risk-stratification systems for ET and PV and their shortcomings
Survey the currently available cytoreductive agents and review their clinical efficacy and adverse effects
Develop a personalized method for determining when to initiate cytoreductive therapy in PV and ET and selecting an appropriate agent
CLINICAL CASE
A 61-year-old woman was incidentally noted to have a platelet count of 851 × 109/L. The rest of her complete blood count was normal. She reported feeling well, overall, and exercised regularly. Her examination was notable for a lack of splenomegaly. A further laboratory evaluation, including iron studies, was normal. A bone marrow (BM) biopsy was performed, demonstrating megakaryocytic hyperplasia with hyperlobulation and an absence of reticulin fibrosis consistent with a diagnosis of essential thrombocythemia (ET). Cytogenetic analysis revealed a 46,XX karyotype, and sequencing uncovered a type 1 CALR mutation at a variant allele frequency of 29%.
Introduction
ET and PV are myeloproliferative neoplasms (MPNs) characterized by the overproduction of platelets and red blood cells, respectively.1 These proliferative MPNs are clinically and morphologically distinct, with PV associated with an expanded red cell mass and frequently leukocytosis and thrombocytosis, while thrombocytosis is typically the sole hematologic abnormality in ET. It is paramount to establish a correct diagnosis in order to guide risk-stratification and therapeutic intervention (Table 1). These diseases share a propensity toward thrombosis, which is the leading cause of morbidity and mortality.2,3 The primary tools for reducing the burden of thrombotic events are medications to lower blood counts, termed cytoreductive therapies. While published guidelines by the NCCN and European LeukemiaNet (ELN) can be useful in determining the timing and choice of cytoreductive therapy, these judgments are nuanced and require the consideration of multiple factors.4-6
2022 International Consensus Classification diagnostic criteria for PV and ET
| Essential thrombocythemia | Polycythemia vera |
|---|---|
| Major criteria | |
| 1. Platelet count ≥450 × 109/L | 1. Elevated hemoglobin concentration or elevated hematocrit or increased red blood cell massf |
| 2. BM biopsy showing proliferation mainly of the megakaryocytic lineage, with increased numbers of enlarged, mature megakaryocytes with hyperlobulated staghorn-like nuclei, infrequently dense clustersa; no significant increase or left shift in neutrophil granulopoiesis or erythropoiesis; no relevant BM fibrosisb | 2. Presence of JAK2 V617F or JAK2 exon 12 mutationg |
| 3. Diagnostic criteria for BCR-ABL1-positive CML, PV, PMF, or other myeloid neoplasms are not met | 3. BM biopsy showing age-adjusted hypercellularity with trilineage proliferation (panmyelosis), including prominent erythroid and granulocytic and increased pleomorphic, mature megakaryocytes without atypia |
| 4. JAK2, CALR, or MPL mutationc | |
| Minor criterion | |
| Presence of a clonal markerd or absence of evidence of reactive thrombocytosise | Subnormal serum erythropoietin level |
| The diagnosis of ET requires either all 4 major criteria or the first 3 major criteria plus the minor criterion | The diagnosis of PV requires either all 3 major criteria or the first 2 major criteria plus the minor criterionh |
| Essential thrombocythemia | Polycythemia vera |
|---|---|
| Major criteria | |
| 1. Platelet count ≥450 × 109/L | 1. Elevated hemoglobin concentration or elevated hematocrit or increased red blood cell massf |
| 2. BM biopsy showing proliferation mainly of the megakaryocytic lineage, with increased numbers of enlarged, mature megakaryocytes with hyperlobulated staghorn-like nuclei, infrequently dense clustersa; no significant increase or left shift in neutrophil granulopoiesis or erythropoiesis; no relevant BM fibrosisb | 2. Presence of JAK2 V617F or JAK2 exon 12 mutationg |
| 3. Diagnostic criteria for BCR-ABL1-positive CML, PV, PMF, or other myeloid neoplasms are not met | 3. BM biopsy showing age-adjusted hypercellularity with trilineage proliferation (panmyelosis), including prominent erythroid and granulocytic and increased pleomorphic, mature megakaryocytes without atypia |
| 4. JAK2, CALR, or MPL mutationc | |
| Minor criterion | |
| Presence of a clonal markerd or absence of evidence of reactive thrombocytosise | Subnormal serum erythropoietin level |
| The diagnosis of ET requires either all 4 major criteria or the first 3 major criteria plus the minor criterion | The diagnosis of PV requires either all 3 major criteria or the first 2 major criteria plus the minor criterionh |
Three or more megakaryocytes lying adjacent without other BM cells in between; in most of these rare clusters, 6 or fewer megakaryocytes may be observed. An increase in huge clusters (>6 cells) accompanied by granulocytic proliferation is a morphological hallmark of pre-PMF.
Very rarely, a minor increase in reticulin fibers may occur at initial diagnosis (grade 1).
It is recommended that highly sensitive assays be used for JAK2 V617F (sensitivity level <1%) and CALR and MPL (sensitivity level 1% to 3%); in negative cases, consider a search for noncanonical JAK2 and MPL mutations.
Assessed by cytogenetics or sensitive next-generation sequencing techniques.
Reactive causes of thrombocytosis include a variety of underlying conditions like iron deficiency, chronic infection, chronic inflammatory disease, medication, neoplasia, or history of splenectomy.
Diagnostic thresholds: hemoglobin level above 16.5 g/dL in men and 16.0 g/dL in women; hematocrit above 49% in men and 48% in women; red blood cell mass 25% above mean normal predicted value.
A BM biopsy may not be required in patients with sustained absolute erythrocytosis (hemoglobin concentrations above 18.5 g/dL in men or 16.5 g/dL in women and hematocrit values above 55.5% in men or 49.5% in women) and the presence of a JAK2 V617F or JAK2 exon 12 mutation.
Highly sensitive assays for JAK2 V617F (sensitivity level <1%) are recommended; in negative cases, consider searching for noncanonical or atypical JAK2 mutations in exons 12 to 15.
CML, chronic myelogenous leukemia; PMF, primary myelofibrosis.
This review focuses on decisions regarding cytoreduction in ET and PV. Other management considerations, including therapeutic phlebotomy in PV, antiplatelet treatment, and anticoagulation in patients who have experienced a thrombosis, are essential but outside the scope of this monograph, as is novel therapeutic development.
Risk stratification in ET and PV
It is imperative to determine which patients are at higher thrombotic risk and may benefit from cytoreductive therapy. Conventional ELN risk-stratification for both ET and PV defines high-risk patients as those who are older than 60 years of age and/or have experienced a prior thrombosis.5,7,8 The latter is a strong predictor of subsequent thrombotic events,9,10 but in the absence of a prior thrombotic event, advanced age has also shown to be a predictor of thrombosis. Specifically, patients with low-risk PV have approximately 2.5 thrombotic events per 100 persons per year compared to 10.9 events per 100 persons per year in high-risk disease.9,11 However, these studies ignore functional status and comorbidities as key contributors toward “biological age” rather than “chronological age.” Clearly, someone who is 60 years and 364 days old will not have a different risk profile the next day. Therefore, I emphasize a complete evaluation for thrombotic risk-stratification, including assessment of cardiovascular and functional status.
ET has several additional thrombosis risk models that incorporate risk factors, including cardiovascular comorbodities and the presence of the JAK2 mutation.12-14 These risk factors have been used to derive the International Prognostic Score of Thrombosis in World Health Organization—Essential Thrombocythemia (IPSET-thrombosis) score that was later revised (Figure 1).10,15 IPSET-thrombosis low-, intermediate-, and high-risk patients have a thrombotic risk of 1.03%, 2.35%, and 3.56% per year.10 Additional models have been developed to predict survival, including the MIPSS-ET and -PV scoring systems.16,17 However, these models have not been calibrated to predict thrombosis and should not be used to determine appropriate patients for cytoreductive therapy.
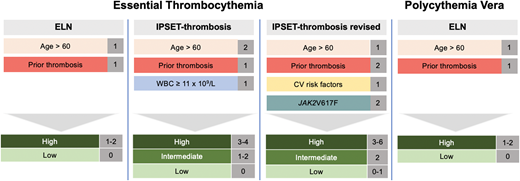
Risk-stratification for thrombosis in ET and PV. In ET there are 3 prognostic models to identify patients at low, intermediate, or high risk of thrombosis, while in PV there is only the European LeukemiaNet risk-stratification system.
Risk-stratification for thrombosis in ET and PV. In ET there are 3 prognostic models to identify patients at low, intermediate, or high risk of thrombosis, while in PV there is only the European LeukemiaNet risk-stratification system.
I advocate the use of the revised IPSET-thrombosis score for ET and ELN risk-stratification for PV. In general, patients who are high risk should be treated with cytoreductive therapy. In terms of intermediate-risk ET patients (eg, age ≥60 with cardiovascular risk factors but without JAK2 mutation or prior thrombosis), I aggressively manage modifiable risk factors and only provide cytoreductive therapy in the setting of vasomotor symptoms or acquired von Willebrand syndrome (aVWS), as described below.
The role of cytoreductive therapy in low-risk ET and PV
Some additional considerations for cytoreduction are outside thrombotic risk status. In ET, and occasionally in PV, patients with extreme thrombocytosis (ie, platelet count >1000 × 109/L) are at risk of developing aVWS. In the case of bleeding from aVWS, cytoreductive therapy should be instituted to reduce the platelet number. In PV, cytoreduction can be utilized to reduce phlebotomy burden in patients who are intolerant of venesection or where it negatively impacts quality of life, although there is no specific number of phlebotomies to prompt the initiation of cytoreductive therapy in low-risk disease.18 The recognition of symptoms as a result of iron deficiency, including fatigue and impaired concentration, is also emerging.19 Other hematologic findings have also been proposed as indications for cytoreductive therapy, most notably leukocytosis in the case of PV. At present, I do not routinely cytoreduce for leukocytosis in the absence of other high-risk features, as an elevated white blood cell (WBC) has not consistently been demonstrated to be of thrombotic prognostic significance.20-22
An additional consideration for cytoreduction in low-risk patients is the presence of certain symptoms, particularly those that are microvascular.23 Splenomegaly, although not frequently observed in ET and PV and typically mild,16,24 can lead to early satiety, and the janus kinase (Jak) inhibitor ruxolitinib effectively ameliorate these symptoms.25 Particularly in ET, where ruxolitinib is not approved, the development of splenomegaly should prompt a BM evaluation to exclude progression to myelofibrosis. Similarly, pruritus is particularly sensitive to treatment with Jak inhibitors.25 Ropeginterferon-α-2b (ROPEG-IFN) has been explored in low-risk PV patients and has demonstrated a reduction in pruritus and night sweats, although with increased fatigue compared to standard treatment.26 As such, I typically do not offer ROPEG-IFN to low-risk patients except to manage specific disease-related symptoms, although I do consider it in low-risk PV patients with phlebotomy intolerance, symptomatic splenomegaly, or progressive thrombocytosis or leukocytosis as recommended by the latest National Comprehensive Cancer Network (NCCN) guidelines.4
Importantly, noncytoreductive therapies for symptoms should be optimized (eg, aspirin for erythromelalgia) before initiating cytoreduction in a low-risk, symptomatic patient. Additionally, cytoreductive therapy for symptom control in ET and PV is most effective in patients with a high symptomatic burden, while minimally symptomatic patients may actually experience worsening symptoms, likely related to adverse events (AEs).27
CLINICAL CASE (continued)
Our patient was deemed to have low thrombotic risk by IPSET-thrombosis, and cytoreductive therapy was not recommended. After approximately 1 year, she developed worsening thrombocytosis to 1263 × 109/L, associated with gum bleeding. She was diagnosed with aVWS, so cytoreductive therapy was reconsidered.
Cytoreductive therapies in ET
Currently available cytoreductive options for appropriate ET patients include hydroxyurea (HU), interferons, anagrelide, and busulfan. Starting with HU, its use is supported by randomized clinical trial data in high-risk patients.28 HU has also been evaluated in lower-risk patients 40 to 59 years of age without a prior thrombosis and failed to show a significant reduction in thrombosis, bleeding, or death,29 highlighting that cytoreductive therapy should only be offered in patients with a high risk of thrombosis.
Pegylated interferon (PEG-IFN) has been thoroughly evaluated for ET treatment both as an initial cytoreductive therapy and after HU intolerance/resistance. Data from the MPN-RC 111, which evaluated PEG-IFN after HU failure in both PV and ET, demonstrated an overall hematologic response rate of 69% among ET patients. Response rates were higher in CALR- mutated patients.30 In the frontline setting in ET, the choice of HU vs PEG-IFN has been informed by several pivotal trials (Table 2). The first is the MPN-RC 112 trial, which randomized newly diagnosed high-risk PV and ET patients to HU or PEG-IFN. The primary end point of a complete response by ELN criteria at 12 months was similar in both treatment arms, as was spleen size reduction and WBC response. Similarly, there was no difference in symptom response between the HU (43%) and PEG-IFN (49%) groups. In the overall cohort (including PV), significantly more patients had a histopathologic response in the HU arm. Reductions in the JAK2 mutational allele burden regardless of treatment group were seen at 12 months, although there was a continued decrease with PEG-IFN, while the allele burden increased after 12 months in the HU arm. Notable toxicity differences included more grade 1 and 2 depression in PEG-IFN–treated patients compared with the HU arm. Other PEG-IFN–related AEs were common, including flu-like symptoms, injection-site reactions, and peripheral sensory neuropathy. Mucositis and anorexia were significantly more common in the HU arm.31 Of note, the number of thrombotic and disease progression events were low, limiting the ability to detect differences between these outcomes.
Summary of randomized trials of hydroxyurea and interferon in ET and PV
| Trial | Patients | Arms | N | CHR at 24 months | Discontinuation rate because of AEs | Additional comments |
|---|---|---|---|---|---|---|
| MPN-RC 11231 | High-risk ET (n = 81) or PV (n = 87) | HU | 80 | 20% | 11% | PEG-IFN led to greater reduction in JAK2V617F; HU had more histopathologic responses |
| PEG-IFN | 82 | 29% | 15% | |||
| DALIAH33,44 | Newly diagnosed ET, PV, pre-PMF, or PMF | HU | 38 | 21% | 13% | Median JAK2V617F reduction was greater at 36 months in PEG-IFN arm compared to HU |
| PEG-IFNa | 164 | 26% | 34% | |||
| PROUD-PV/ CONTINUATION-PV45 | High-risk PV untreated or <3 years of HU | HU | 76 | 49% | 4% | Molecular responses were higher in the ROPEG-IFN arm compared to standard therapy at 24 and 36 months |
| ROPEG-IFN | 95 | 71% | 8% |
| Trial | Patients | Arms | N | CHR at 24 months | Discontinuation rate because of AEs | Additional comments |
|---|---|---|---|---|---|---|
| MPN-RC 11231 | High-risk ET (n = 81) or PV (n = 87) | HU | 80 | 20% | 11% | PEG-IFN led to greater reduction in JAK2V617F; HU had more histopathologic responses |
| PEG-IFN | 82 | 29% | 15% | |||
| DALIAH33,44 | Newly diagnosed ET, PV, pre-PMF, or PMF | HU | 38 | 21% | 13% | Median JAK2V617F reduction was greater at 36 months in PEG-IFN arm compared to HU |
| PEG-IFNa | 164 | 26% | 34% | |||
| PROUD-PV/ CONTINUATION-PV45 | High-risk PV untreated or <3 years of HU | HU | 76 | 49% | 4% | Molecular responses were higher in the ROPEG-IFN arm compared to standard therapy at 24 and 36 months |
| ROPEG-IFN | 95 | 71% | 8% |
PEG-IFN included both interferon-α-2a and interferon-α-2b.
PMF, primary myelofibrosis; PR, partial response.
The DALIAH trial randomized untreated MPN patients, including 73 ET patients, to either HU, PEG-IFN, or PEG-IFN-α-2b. Eligible patients had active disease as evidenced by the need for therapeutic phlebotomy, a WBC count above 10 × 109/L, a platelet count higher than 400 × 109/L, constitutional symptoms, pruritus, symptomatic splenomegaly, or previous thrombosis. At 36 months the overall hematologic response rate was higher in the HU group vs the combined interferon groups. In addition, treatment discontinuation because of AEs was significantly higher for interferon-treated patients. The median JAK2 V617F allele burden reduction was higher in the interferon arms.32 The CALR variant allele frequency did not significantly decline, and surprisingly, there were more treatment-emergent DNMT3A mutations in the interferon therapy arm compared with HU, suggesting that interferon does not prevent molecular evolution.33
Taken together, the MPN-RC 112 and DALIAH trials highlight that both HU and PEG-IFN are active treatments for ET and that either can be used as frontline cytoreductive therapy. In practice I advocate for a thorough discussion about toxicity profiles, about expected hematologic outcomes, and about the lack of substantial data on the differences in thrombosis or progression rates when discussing the difference between these agents.
In ET, another cytoreductive therapy option is anagrelide, an oral imidazoquinazoline derivative that represses megakaryocytic differentiation and that is dosed initially at 0.5 mg twice daily and up-titrated weekly by 0.5 mg based on hematologic response and tolerance.34 Anagrelide has also been compared to HU as a first-line cytoreductive therapy. In a PT-1 study, patients with high-risk ET were randomized to either HU or anagrelide. During follow-up, anagrelide-treated patients were significantly more likely to experience thrombosis, hemorrhage, or death. Anagrelide treatment was associated with an increased rate of arterial thrombosis, hemorrhage, and transformation to myelofibrosis but a decreased rate of venous thrombosis.35 Because the PT-1 trial used antiquated ET diagnostic criteria, the ANAHYDRET study was performed to assess whether anagrelide was noninferior to HU in untreated ET patients deemed to have high-risk factors, defined as aged 60 years or older, a platelet count equal to or greater than 1000 × 109/L, an increase in platelet count of more than 300 × 109/L within 3 months, cardiovascular risk factors including hypertension and diabetes, and/or a history of a thrombohemorrhagic event. There was no difference in the primary end point of thrombosis or bleeding, meeting its prespecified criteria for noninferiority. Cardiovascular side effects were more frequent in the anagrelide treatment arm, while leukopenia and minor infections were more common with HU.36 Although an effective agent to lower platelet counts, dosing is often limited by toxicity, including palpitations/tachycardia, fluid retention, and diarrhea.37
Cytoreductive therapies in PV
The choice for first-line cytoreductive therapy in PV includes both HU and PEG-IFN, while ruxolitinib can be given in the setting of HU resistance or intolerance. HU use in high-risk PV is supported by retrospective data demonstrating a reduction in thrombosis,38 although prospective randomized data demonstrating improvement in thrombotic burden with HU treatment are lacking. PEG-IFN has been explored in the treatment of PV in the MPN-RC 111 trial, which found an overall hematologic response rate at 12 months of 60%.30 PEG-IFN and ROPEG-IFN are also an appropriate choice for frontline cytoreductive therapy as endorsed by NCCN guidelines.4
Ruxolitinib is approved for the treatment of PV in patients resistant or intolerant to HU based on the RESPONSE trial, which randomized PV patients with palpable splenomegaly to either ruxolitinib or best available therapy. Hematocrit control was significantly improved in ruxolitinib-treated patients. Importantly, ruxolitinib led to significant improvements in symptoms, with particularly striking reductions in pruritus, night sweats, and early satiety.25 These findings were also echoed in the PV population without splenomegaly in a subsequent trial.39 In the recently published MAJIC-PV study, which randomized HU-intolerant or -resistant PV patients to either best available therapy or ruxolitinib, there was increased duration of response, improvement in event-free survival (thrombosis, hemorrhage, transformation, and death), and a significant increase in molecular responses observed with ruxolitinib therapy.40 Ruxolitinib is a particularly attractive cytoreductive agent for patients with considerable symptom burden, particularly pruritus, and may reduce thrombotic burden.40,41
Another cytoreductive agent for both ET and PV is the alkylating agent busulfan, which is given at a starting dose of 2 mg/d. This agent can induce hematologic responses and can be given intermittently when counts rise.42 However, it has been associated with an increased risk of leukemia, and therefore its use should be limited to elderly patients who have failed or are intolerant to other cytoreductive agents.43
Similar to ET, multiple investigations have compared first-line cytoreductive therapies in PV (Table 2). The previously discussed MPN-RC 112 trial showed that the overall response rate (ORR) at 12 months was not statistically different in the PV patient population.31 The DALIAH trial also included a cohort of PV patients. There was no significant difference in overall response rates (ORRs) between the HU and interferon groups; however, maintenance of complete hematologic response was longer, and a reduction in the JAK2 V617F allele was greater with interferon compared to HU.44
A monopegylated formulation of interferon-α-2b, ROPEG-IFN, has been developed that allows for every-other-week dosing. It is approved for the treatment of PV based on the PROUD-PV/ CONTINUATION-PV study, which enrolled patients who were treatment naive or previously treated for less than 3 years with HU. High-risk patients were defined as aged older than 60 years or having a prior thrombotic event, phlebotomy intolerance, progressive splenomegaly, a platelet count above 1000 × 109/L, or a leukocyte count higher than 10 × 109/L. Importantly, these inclusion criteria are different than the MPN-RC 112 trial, most notably the allowable duration of HU exposure before enrollment (3 years vs 3 months), limiting comparisons. There was no significant difference in complete hematologic response (CHR) at 12 months between ROPEG-IFN and HU. At 36 months, the end point of CHR plus improved disease burden (defined as resolution or clinical improvement of disease-related splenomegaly, microvascular disturbances, pruritus, or headache) was higher in the ROPEG-IFN arm. Molecular responses were significantly higher with HU at 12 months but became higher at 24 and 36 months in the ROPEG-IFN arm. There was no significant difference in thrombosis rates between the two arms.45 Although it is tempting to correlate the reduction in JAK2 mutational burden with a decreased rate of progression, there is insufficient evidence at present to support this claim. Of note, retrospective evidence suggests that interferon-based therapies are associated with a decreased myelofibrosis-free and overall survival.46 Although provocative, given the inherent limitations with this study design, these findings need to be confirmed in larger, ideally prospective investigations before they can be incorporated into clinical practice.
The decision to use ROPEG-IFN vs HU for cytoreduction in PV is largely redundant with PEG-IFN vs HU. If an interferon-based approach is determined, I will employ ROPEG-IFN given its dosing schedule and full regulatory approval for the treatment of PV.
Assessing response (focus on hematologic parameters)
Once a cytoreductive strategy has been chosen, how do you determine a response? ELN criteria for both ET and PV dictate the normalization of blood counts, in addition to a lack of thrombosis, bleeding, or disease progression to myelofibrosis.47 However, these response criteria have not been validated, and in fact several lines of retrospective evidence suggest that obtaining an ELN response in ET and PV is not associated with a decreased risk of thrombosis or survival (Table 3).48-51 As the primary function of these response criteria is for uniform reporting of clinical trial results, increasing the dose of cytoreductive therapy or changing treatment to achieve a response is not recommended. In ET the optimal platelet goal is not established, and the fact that there is no clear relationship between platelet count and thrombosis risk is well recognized.52,53
Studies assessing the association of ELN response and thrombosis or survival
I typically aim to control platelet count to a near-normal range without compromising tolerability. In particular, if the normalization of platelet count results in symptomatic anemia or moderate neutropenia, then the dose of a cytoreductive agent should be reduced. In PV patients being treated with cytoreductive therapy who require therapeutic phlebotomy, I titrate the dose to limit phlebotomies to less than 3 annually, as this has been associated with a decreased risk of thrombotic complications (albeit only in the context of HU treatment).54 Importantly, interferon-based therapies may take longer to achieve a response.45 I typically increase the dose monthly to optimize hematologic control and other treatment goals. Most importantly, it is key to continually assess changes in symptoms, both in terms of improvements as well as the emergence of therapy-related AEs.
Special circumstances
In selected circumstances there is a clear preference for one cytoreductive therapy over another. In the setting of pregnancy and in patients trying to conceive, PEG-IFN has demonstrated safety based on retrospective series,55,56 although available data are not robust enough to exclude safety concerns as they relate to fetal development. In contrast, HU is a known teratogen. Therefore, in younger patients who are considering conception I recommend interferon-based therapies. There has been a historic concern that HU is leukemogenic; however, contemporary analysis has not substantiated this association.57 Interferons should not be given in the setting of severe depression, anxiety, or other psychiatric conditions or in patients with acute/untreated autoimmune disease.58 Anagrelide should be used with caution in patients with congestive heart failure or arrhythmias.
Conclusions
As described above, the choices around who, what, when, and how to deliver cytoreductive therapy in ET and PV are highly individualized and should involve the consideration of multiple factors, including patient preference. To date, there is insufficient evidence to suggest that there are clinically significant efficacy advantages to one cytoreductive therapy over another. Ideally, prospective clinical trials would be able to discriminate thrombosis rates, time to disease progression, and ultimately survival. However, these events occur over the course of decades at relatively low rates, requiring large and long clinical trials that are not feasible. Targeting high-risk subjects to increase the expected event rate should be considered when designing studies with thrombotic end points, in particular.50 Biomarkers and additional clinical features may also serve as surrogate end points in the future, although data to identify and validate these metrics are lacking. In the future, with the development of novel therapies for ET and PV that can not only reduce thrombotic burden but also modify disease progression and survival, the use of risk scores that predict survival (eg, MIPSS-ET and -PV) may be relevant for deciding when and how to initiate cytoreductive therapy. Increased recognition and attention toward the identification/validation of surrogate end points for thrombosis and the development of novel therapeutics will be required to advance ET and PV clinical research and identify the optimal cytoreductive strategy in these MPNs.
Acknowledgments
The author would like to thank Ronald Hoffman, John Mascarenhas, and Marina Kremyanskaya for their expert review of the manuscript.
Conflict-of-interest disclosure
Douglas Tremblay: research funding: CTI Biopharma, Astellas Pharma, Gilead; consultancy: CTI Biopharma, Novartis, AbbVie, Sierra Oncology, GSK, Cogent Biosciences.
Off-label drug use
Douglas Tremblay: pegylated interferon
