

Abstract
Among the variety of resistance mechanisms that may underlie a non-optimal response to tyrosine kinase inhibitor (TKI) therapy in chronic myeloid leukemia patients, secondary point mutations in the BCR::ABL1 kinase domain (KD) represent the only actionable one. Each of the 5 ATP-competitive inhibitors (imatinib, dasatinib, nilotinib, bosutinib, ponatinib) has a well-defined spectrum of resistance mutations. Growing clinical experience will soon allow to also elucidate the full spectrum of mutations conferring resistance to asciminib (that appear not to be confined to the myristate binding pocket). Regular molecular response (MR) monitoring is fundamental for evaluating treatment efficacy, catching early signs of relapse, and intervening promptly in case of confirmed failure. Whenever MR is not deemed satisfactory according to the European LeukemiaNet or the National Comprehensive Cancer Network definitions, BCR::ABL1 KD mutations testing should be performed. When needed, prompt and informed TKI switch can improve response and outcome and prevent the accumulation of mutations, including highly challenging compound mutations. Novel technologies like next-generation sequencing and digital polymerase chain reaction have recently been explored for BCR::ABL1 KD mutation testing; they have both advantages and disadvantages that are discussed in this article. This review also provides suggestions for interpretation and clinical translation of mutation testing results, which may not always be straightforward, particularly in cases of low-level or unknown mutations.
Learning Objectives
Identify chronic myeloid leukemia patients who need testing for resistance mutations
Weigh the role of BCR::ABL1 mutation status in clinical decision-making
Introduction
Selection of point mutations in the kinase domain (KD) of BCR::ABL1 can be observed in chronic myeloid leukemia (CML) patients who relapse on tyrosine kinase inhibitor (TKI) therapy or who do not achieve the target response. BCR::ABL1 KD mutations are not the most frequent mechanism of resistance to therapy—yet they remain the only actionable one.
CLINICAL CASE
A 62-year-old man is diagnosed with chronic phase (CP)-CML. BCR::ABL1 transcript type is e13a2; both Sokal and EUTOS long-term survival score are intermediate. The patient is started on imatinib 400 mg/d. Monitoring of BCR::ABL1 transcript levels by reverse transcription–quantitative polymerase chain reaction on peripheral blood yields the following results, expressed on the International Scale (IS):
Baseline: 88%IS
3 months: 12%IS
6 months: 8.7%IS
9 months: 6.1%IS
12 months: 3.8%IS
How would you manage this patient?
When should testing for BCR::ABL1 KD mutations be performed?
The clinical value of BCR::ABL1 KD mutations testing is widely recognized, so that both the European LeukemiaNet (ELN)1 and the National Comprehensive Cancer Network (NCCN)2 recommend testing when response is not satisfactory. Both the ELN and NCCN base the evaluation of response on the stepwise achievement of key molecular response (MR) milestones at given checkpoints during therapy, with some slight differences in timing and levels. The ELN distinguishes nonoptimal responses into “failures” (therapy must be changed) and “warnings” (“. . . continuation or change should be carefully considered, depending on patient's characteristics, comorbidities and tolerance as well as on therapeutic endpoints”), while NCCN uses a traffic-light approach, with red corresponding to TKI-resistant disease and yellow corresponding to possible TKI resistance. In case of failure/red or warning/yellow, which mean that the expected MR milestone has not been achieved, or when a previously achieved milestone is lost, thorough investigation into the underlying reason(s) should be undertaken, bearing in mind that an unsatisfactory response may be due to reduced compliance, drug interactions (especially in elderly patients taking many concomitant medications), or truly resistant disease.
Reliable molecular monitoring by reverse transcription–quantitative polymerase chain reaction, performed regularly (every 3 months until major molecular response [MMR] is achieved and confirmed, and every 3 to 6 months thereafter)1 is instrumental to check MR for evaluating treatment efficacy, to catch early signs of relapse, to trigger mutation testing when appropriate, and to intervene quickly in case of manifested failure. BCR::ABL1 kinase activity is thought to feed genetic instability, thus incomplete or inefficient BCR::ABL1 inhibition is insidious in that it might set the ground for acquiring mutations in the KD or elsewhere in the genome and for additional cytogenetic abnormalities. Beyond a certain threshold, accumulating genetic lesions may ultimately lead to disease progression, which remains a major concern and must be avoided.3 The kinetics of BCR::ABL1 increase (“doubling time”) may help identify both disease recurrence in case of non-adherence and impending resistance due to mutation development. Shorter doubling times (median = 9 days) have been associated with the former, whereas longer doubling times (median = 48 days) have been associated with the latter.4 However, the study was conducted using BCR as a control gene; validating these observations using the other control genes usually employed—ABL1 and GUSB—would help incorporate the assessment of transcript kinetics into routine use.
BCR::ABL1 KD mutation testing is not recommended at diagnosis. Even when using a highly sensitive and reliable approach of single molecule sequencing, mutations could not be detected in de novo CP patients.5 Hence, more “routine” methods would inexorably yield negative results. This is consistent with the fact that mutant clones require the selective pressure of treatment to outgrow: without such pressure, they are very unlikely to outcompete the unmutated clone. Accordingly, patients who lose MMR after attempting to discontinue treatment have not been reported to harbor mutations. For this same reason, samples for mutation testing should be taken before stopping or switching therapy in order to not modify the selective pressure—particularly when using Sanger sequencing, which, despite its limitations, remains the most widely employed method for testing, as discussed below.
Last but not least, we must bear in mind that mutation testing is technically feasible (regardless of the method used) only if BCR::ABL1 transcript levels are >0.1% (that is, the threshold of MMR), and that reliability and reproducibility of results improve when the transcript levels are >1%.6 Thus, patients in MMR (or better) should not be tested for mutations: this would not make sense clinically and would waste efforts and resources for the laboratory. A practical algorithm that can be used to assess whether mutation testing is advisable is shown in Figure 1.
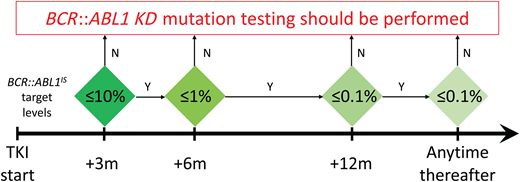
Molecular response milestones to be achieved and maintained in first- and second-line TKI therapy for optimal response to be defined according to the ELN. BCR::ABL1 KD mutation testing may provide useful information if such milestones are missed. KD, kinase domain; m, months; TKI, tyrosine kinase inhibitor.
Molecular response milestones to be achieved and maintained in first- and second-line TKI therapy for optimal response to be defined according to the ELN. BCR::ABL1 KD mutation testing may provide useful information if such milestones are missed. KD, kinase domain; m, months; TKI, tyrosine kinase inhibitor.
CLINICAL CASE (continued)
The patient has failed all the MR milestones set to be achieved during the first 12 months of treatment. The patient swears he has been taking imatinib regularly. His physician ultimately sends a peripheral blood sample for BCR::ABL1 KD mutation testing, but Sanger sequencing shows no evidence of mutations. At month +15 (BCR::ABL1 = 4.2%IS) the patient is switched to nilotinib 400 mg/twice a day, but MR does not improve: BCR::ABL1 levels slowly but steadily increase to 7.2% at 18 months, 8.5% at 21 months, and 10.1% at 24 months. Sanger sequencing is repeated and an E255K mutation is detected. The patient is then switched to dasatinib 140 mg/daily, but he progresses to blast crisis after 3 months. BCR::ABL1 KD mutation testing shows evidence of an E255K mutation and a T315I mutation, both at 100%.
Compound mutations: the ultimate enemy
Each of the 5 ATP-competitive inhibitors (imatinib, dasatinib, nilotinib, bosutinib, ponatinib) have a well-defined spectrum of resistance mutations. Ponatinib is efficacious against every individual mutation among those conferring resistance to imatinib, dasatinib, nilotinib, and bosutinib, but its activity is compromised when a further nucleotide substitution at codon 315 turns a preexisting T315I into a T315M or -L mutation.
More recently asciminib, an allosteric inhibitor specifically targeting the myristate-binding pocket rather than the ATP-binding pocket, has been approved for the treatment of patients resistant to at least 2 previous TKIs. Given that asciminib binds to a region different and distant from the region targeted by ATP-competitive inhibitors, the molecule was initially predicted to have a nonoverlapping spectrum of resistance mutations.7 However, accumulating evidence from clinical trials indicates that even well-known imatinib- or 2G TKI-resistant mutations like E355G and F359V may be identified at the time of asciminib failure (Table 1). Yet mutation data from trials are scarce, and “real-life” clinical experience with asciminib is still limited. Thus, time is needed before the full spectrum of asciminib-resistant mutations will be conclusively elucidated.
Provisional list of mutations that might confer resistance to asciminib based on the currently available data
| Reference | Type of study | Mutations |
|---|---|---|
| Wylie et al. (2017)7 | In vitro screen in KCL-22 CML cells cultured in increasing concentrations of asciminib and IC50 data in luciferase-transformed murine Ba/F3 cells expressing native BCR::ABL1 as compared with various BCR::ABL1 mutants | P223S; K294E; A337V; P465S; V468F; I502L |
| Qiang et al. (2017)28 | In vitro screen of asciminib-resistant CML cell lines cultured in increasing concentrations of asciminib | C464W; M244V/A337V |
| Eide et al. (2019)11 | In vitro cell-based accelerated mutagenesis screen and IC50 data in murine Ba/F3 cells expressing native BCR::ABL1 as compared with various BCR::ABL1 mutants | A344P; F359V; F359I; F359C; P465S; G671R (based on outgrowing mutants) T315L; T315M; T315I/G250E; T315I/Y253H; T315I/E255V; T315I/M351T; T315I/H386R; T315I/E453K; E255V/299L; V299L/F317L; F317L/F359V (based on IC50 data in a selected panel of single mutations and CMs of interest) |
| Hughes et al. (2019)29 | Clinical trial (phase 1, CP and AP) | G109D*; Y115N*; A337T; G463S; G463D*; P465S*; V468F; I502L |
| Mauro et al. (2023)30 | Clinical trial (phase 1; 4-year update of T315I-negative CP patients) | M244V; G463D; G463S; V468F; I502L |
| Hochhaus et al. (2023)31 | Clinical trial (phase 3 asciminib vs bosutinib [ASCEMBL]) | M244V; E355G; F359V; T315I; A337T; P465S |
| Cortes et al. (2020)32 (ASH meeting abstract) | Clinical trial (phase 1, T315I-positive patients) | F359I; T315I/F359I; A337T/F359V; T315I/M244V; T315I/M351T; T315I/E453Q |
| Reference | Type of study | Mutations |
|---|---|---|
| Wylie et al. (2017)7 | In vitro screen in KCL-22 CML cells cultured in increasing concentrations of asciminib and IC50 data in luciferase-transformed murine Ba/F3 cells expressing native BCR::ABL1 as compared with various BCR::ABL1 mutants | P223S; K294E; A337V; P465S; V468F; I502L |
| Qiang et al. (2017)28 | In vitro screen of asciminib-resistant CML cell lines cultured in increasing concentrations of asciminib | C464W; M244V/A337V |
| Eide et al. (2019)11 | In vitro cell-based accelerated mutagenesis screen and IC50 data in murine Ba/F3 cells expressing native BCR::ABL1 as compared with various BCR::ABL1 mutants | A344P; F359V; F359I; F359C; P465S; G671R (based on outgrowing mutants) T315L; T315M; T315I/G250E; T315I/Y253H; T315I/E255V; T315I/M351T; T315I/H386R; T315I/E453K; E255V/299L; V299L/F317L; F317L/F359V (based on IC50 data in a selected panel of single mutations and CMs of interest) |
| Hughes et al. (2019)29 | Clinical trial (phase 1, CP and AP) | G109D*; Y115N*; A337T; G463S; G463D*; P465S*; V468F; I502L |
| Mauro et al. (2023)30 | Clinical trial (phase 1; 4-year update of T315I-negative CP patients) | M244V; G463D; G463S; V468F; I502L |
| Hochhaus et al. (2023)31 | Clinical trial (phase 3 asciminib vs bosutinib [ASCEMBL]) | M244V; E355G; F359V; T315I; A337T; P465S |
| Cortes et al. (2020)32 (ASH meeting abstract) | Clinical trial (phase 1, T315I-positive patients) | F359I; T315I/F359I; A337T/F359V; T315I/M244V; T315I/M351T; T315I/E453Q |
Mutations have either been recovered from in vitro studies where resistant cell lines were obtained using various strategies or identified as newly emerging mutations in patients enrolled in the clinical trials. Myristate-binding site mutations are highlighted in italic. The asterisk denotes mutations detected in patients with a variant frequency <10%, mostly in combination with other myristate-binding pocket or catalytic site mutations.
While single mutations can be tackled by one or more of the currently available TKIs, compound mutations (CMs; 2 mutations in cis on the same BCR::ABL1 transcript) have emerged as a major concern. Data of in vitro IC50 (ie, the intracellular concentration of drug required to inhibit by 50% the growth of a cell line engineered to express the given mutant oncoprotein), corroborated by an increasing number of clinical reports, indicate that the most frequent mutation combinations (that are usually T315I-inclusive CMs) are resistant to all available TKIs, including, in some cases, even ponatinib and asciminib (Table 1).5,8-11 CMs usually result from sequential TKI failures, although they have also been documented in a few patients whose switch to another TKI was delayed despite persisting failure: if the original mutant clone is not rapidly and fully eradicated, it may acquire a “second hit” that may lead to a highly challenging CM.5,12 At least in vitro, a combination of asciminib and ponatinib at clinically achievable concentrations is capable of counteracting (as well as preventing) several CMs.11,13 Combination of ponatinib with hydroxyurea or with the CDK4/6 inhibitor palbociclib has also shown promising activity in vitro against T315I-inclusive CMs.14 It is likely that other combinations exploiting specific vulnerabilities of CMs or synthetic lethality may be devised in the future. However, given the difficulty of exploring off-label combinations in vivo, the best strategy is currently to prevent, rather than to counteract, CMs.
Sanger sequencing vs newer techniques: pros and cons
Sanger sequencing enables scanning of the entire KD for any nucleotide substitution. Given the multitude of mutations associated with imatinib resistance, it naturally became the gold standard for BCR::ABL1 KD mutation testing15 and remains such after 2 decades. Mutation screening of BCR::ABL1 transcripts by Sanger sequencing is relatively fast and easy but suffers from the inherent poor sensitivity of the technique (ranging between 10% and 20%, that is, 10 to 20 mutant transcripts in 100 total BCR::ABL1 transcripts), meaning the technique can only identify major or dominant mutant clones. It is thus not surprising that, in recent years, several retrospective studies and two prospective studies have explored the use of targeted next generation-sequencing (NGS) that improves the detection limit to 1% to 5%.16-20 These studies have shown that Sanger sequencing may miss minor additional subclones at the time of treatment failure, as well as emerging ones in patients with warning responses. Moreover, NGS may more easily and robustly identify CMs (see below), although it is important to correct results for the likelihood of polymerase chain reaction (PCR)–mediated recombination21 that may artifactually bring in cis 2 mutations sitting on different molecules.22 However, despite wider and wider availability of benchtop NGS instruments, implementing routine NGS for BCR::ABL1 KD mutation testing is facing some challenges. First is the need to set up and internally validate a laboratory-developed test. Second is the high throughput of the instruments as compared with the dimensions of the library, which requires pooling several samples in each sequencing run. Consequently, only sample centralization in regional or national reference laboratories could balance cost-effectiveness and turnaround time. Last but not least of the challenges is the higher error rate as compared with Sanger sequencing.
Mutation-specific approaches taking advantage of mass spectrometry (MS) or digital PCR can be more sensitive and less error-prone than NGS. MS was indeed the first high-sensitivity (0.05% to 0.5%, depending on the mutation) approach to be explored. A multiplex strategy of primer extension followed by MS-based identification of the extended nucleotide developed for a panel of 31 imatinib-resistant mutations was applied to a relatively large cohort of imatinib-resistant patients who were switched to dasatinib or nilotinib.23 The study provided the first robust evidence that detection of low-level mutations at the time of TKI switch may offer critical information to guide subsequent therapy selection, and that if an “inappropriate” TKI is selected, there is a high risk of treatment failure with clonal expansion of the resistant mutant.23 A more recent development is a droplet digital PCR (ddPCR)–based strategy multiplexing the detection and quantitation of 2G TKI-resistant mutations in a 3-tube format.24 The first tube contains primers and probes to detect the T315I mutation; the second tube is specific for dasatinib- and bosutinib-resistant mutations, and the third tube detects nilotinib- and bosutinib-resistant mutations. The appearance of a positive cluster of droplets and where such cluster sits in the 2D plot directly indicate which 2G TKI(s) should be excluded from the decision algorithm at the time of switch (Figure 2). All the approaches discussed so far look for mutations in BCR::ABL1 transcripts, because at the genomic level the selective analysis of the translocated allele would be impossible due to the width of the region to be amplified (17 kb). Nevertheless, an allele- specific ddPCR strategy using genomic DNA to estimate mutated cells and follow their clonal evolution over time has recently been described.25 The approach quantitates the mutated clone as the percent ratio between the copy number of mutated ABL1 and the copy number of the BCR-ABL1 fusion assessed at the DNA level with patient-specific primers. Comparison with NGS conventionally performed using RNA as input nucleic acid showed very good concordance between the level of mutation at the transcript and genomic level, although, as expected, ddPCR featured greater sensitivity. Routinely implementing a DNA-based strategy would be impractical due to the need to determine each patient's exact sequence of the BCR::ABL1 breakpoint at diagnosis, yet this study further highlights the advantages of ddPCR in terms of rapidity and sensitivity.
![Example of a ddPCR-based strategy for detecting 2G TKI-resistant mutations at the time of switch. Representative 2-dimensional plots (in which channel 1 fluorescence [FAM] is plotted against channel 2 fluorescence [HEX]) expected for mutations conferring resistance to one or more 2G TKI. Black clusters at the bottom left corner represent FAM/HEX double-negative droplets. Green clusters at the bottom right corner correspond to droplets positive for e13a2 or e14a2 BCR::ABL1 fusion transcripts (detected by HEX-conjugated probes). Blue clusters at the top left corner correspond to droplets positive for the indicated BCR::ABL1 KD mutations or mutation subgroups (detected by FAM or FAM/HEX-conjugated probes). Orange clusters at the top right corner correspond to double-positive droplets. One tube is specific for the pan-resistant T315I mutation; one tube detects dasatinib-resistant mutations (with the nucleotide substitution leading to the V299L mutation clustering separately from the others since they also confer resistance to bosutinib); and one tube detects nilotinib-resistant mutations (with the E255K mutation clustering separately from the others since it also confers resistance to bosutinib). Results are expressed as percentage of mutant transcripts over total BCR::ABL1 transcripts. 2G TKI, second-generation tyrosine kinase inhibitor.](https://ash.silverchair-cdn.com/ash/content_public/journal/hematology/2023/1/10.1182_hematology.2023000447/2/m_hem2023000447figure2.png?Expires=1767745822&Signature=AYI09gvAG23M~q3PTBzj4HpYYkT74~K8z38eD5uSNBoHJJ7fSqIt8GAP-0eAbzabI5JnOh9YtHCQ4rNaCg9EbmeccZ328LJPLndxVgnhiqPxyBqlj0Fo5IMCpxUyo~UeeqTIWKJHmP18T~LlwCCoHcfUvuPhHGWahdVpM02SlcN9TxlxkivWjvrT6gLgpmQwnYLSBYjI2MZnAqcA0EJ5RJ6djufxTkOJExeJzpVDcO8d9d2zlJ2JyfsOC5MAvC0FU7eXV9R0oIT~azi6IsIEacBtPjdQVUTDWKPKT4fTMOB1dpif7Ocia0tkQwygMIoO4~sTlsfacWh1QXDMJbCnVw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Example of a ddPCR-based strategy for detecting 2G TKI-resistant mutations at the time of switch. Representative 2-dimensional plots (in which channel 1 fluorescence [FAM] is plotted against channel 2 fluorescence [HEX]) expected for mutations conferring resistance to one or more 2G TKI. Black clusters at the bottom left corner represent FAM/HEX double-negative droplets. Green clusters at the bottom right corner correspond to droplets positive for e13a2 or e14a2 BCR::ABL1 fusion transcripts (detected by HEX-conjugated probes). Blue clusters at the top left corner correspond to droplets positive for the indicated BCR::ABL1 KD mutations or mutation subgroups (detected by FAM or FAM/HEX-conjugated probes). Orange clusters at the top right corner correspond to double-positive droplets. One tube is specific for the pan-resistant T315I mutation; one tube detects dasatinib-resistant mutations (with the nucleotide substitution leading to the V299L mutation clustering separately from the others since they also confer resistance to bosutinib); and one tube detects nilotinib-resistant mutations (with the E255K mutation clustering separately from the others since it also confers resistance to bosutinib). Results are expressed as percentage of mutant transcripts over total BCR::ABL1 transcripts. 2G TKI, second-generation tyrosine kinase inhibitor.
Example of a ddPCR-based strategy for detecting 2G TKI-resistant mutations at the time of switch. Representative 2-dimensional plots (in which channel 1 fluorescence [FAM] is plotted against channel 2 fluorescence [HEX]) expected for mutations conferring resistance to one or more 2G TKI. Black clusters at the bottom left corner represent FAM/HEX double-negative droplets. Green clusters at the bottom right corner correspond to droplets positive for e13a2 or e14a2 BCR::ABL1 fusion transcripts (detected by HEX-conjugated probes). Blue clusters at the top left corner correspond to droplets positive for the indicated BCR::ABL1 KD mutations or mutation subgroups (detected by FAM or FAM/HEX-conjugated probes). Orange clusters at the top right corner correspond to double-positive droplets. One tube is specific for the pan-resistant T315I mutation; one tube detects dasatinib-resistant mutations (with the nucleotide substitution leading to the V299L mutation clustering separately from the others since they also confer resistance to bosutinib); and one tube detects nilotinib-resistant mutations (with the E255K mutation clustering separately from the others since it also confers resistance to bosutinib). Results are expressed as percentage of mutant transcripts over total BCR::ABL1 transcripts. 2G TKI, second-generation tyrosine kinase inhibitor.
The main advantages and disadvantages of current technologies for BCR::ABL1 KD mutation testing are summarized in Table 2.
Advantages and disadvantages of the main methods currently available for BCR::ABL1 KD mutation testing
| Method | Lower limit of detection | Mutation-specific? | Advantages | Disadvantages |
|---|---|---|---|---|
| Sanger sequencing | 10%-20% | N | Enables TKD-wide screening; easy workflow and data analysis; relatively short turnaround time | Poorly sensitive |
| Mass spectrometry | 0.05%-0.5% | Y | Accurate; highly sensitive | Not widely available; high throughput; longer turnaround time; no commercial kits available; can be implemented for a limited number of mutations; difficult identification of the T315L and T315M mutations (that are caused by a double nucleotide substitution) |
| NGS | 1%-5% | N | Enables TKD-wide screening; sensitive | High throughput; longer turnaround time; requires specialized personnel and bioinformatic competences; relatively high error rate; no commercial kits available |
| ddPCR | 0.1%-0.5% | Y | Accurate; highly sensitive; relatively easy workflow and data analysis; short turnaround time | Can be implemented for a limited number of mutations |
| Method | Lower limit of detection | Mutation-specific? | Advantages | Disadvantages |
|---|---|---|---|---|
| Sanger sequencing | 10%-20% | N | Enables TKD-wide screening; easy workflow and data analysis; relatively short turnaround time | Poorly sensitive |
| Mass spectrometry | 0.05%-0.5% | Y | Accurate; highly sensitive | Not widely available; high throughput; longer turnaround time; no commercial kits available; can be implemented for a limited number of mutations; difficult identification of the T315L and T315M mutations (that are caused by a double nucleotide substitution) |
| NGS | 1%-5% | N | Enables TKD-wide screening; sensitive | High throughput; longer turnaround time; requires specialized personnel and bioinformatic competences; relatively high error rate; no commercial kits available |
| ddPCR | 0.1%-0.5% | Y | Accurate; highly sensitive; relatively easy workflow and data analysis; short turnaround time | Can be implemented for a limited number of mutations |
ddPCR, droplet digital polymerase chain reaction; N, no; NGS, next generation sequencing; TKD, tyrosine kinase domain; Y, yes.
Despite the limitations highlighted here, Sanger sequencing and NGS are the only ways to obtain a detailed snapshot of BCR::ABL1 mutation status. Thus, they are the most informative methods in (1) imatinib-resistant patients when the causes of an unsatisfactory response are to be investigated, (2) multi-TKI-resistant or advanced-phase patients where two or more mutations (and CMs) can be expected, and (3) patients on asciminib. When a TKI switch is already planned, ddPCR might instead be useful to rapidly inform rational selection of 2G TKIs or ponatinib.
CLINICAL CASE (continued)
The patient provides written informed consent for his samples to be retrospectively reanalyzed by NGS. NGS reveals that the E255K mutation was already detectable in 6.7% and 12.1% of BCR::ABL1 transcripts at 9 and 12 months, respectively. Being E255K resistant to nilotinib, this mutation was selected by subsequent nilotinib treatment. Moreover, NGS showed that the acquisition of the T315I, generating the T315I/E255K CM, occurred during nilotinib treatment (Figure 3).
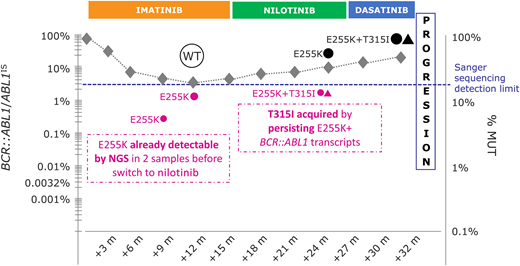
Graphical summary of the clinical case herein presented. m, month(s); % MUT, percentage of mutant; NGS, next-generation sequencing; WT, wild type.
Graphical summary of the clinical case herein presented. m, month(s); % MUT, percentage of mutant; NGS, next-generation sequencing; WT, wild type.
How mutation results should (and shouldn't!) be used
BCR::ABL1 KD mutation testing supports clinical decision-making whenever response is unsatisfactory. In case of failure, a change of therapy is mandatory and the main aim of BCR::ABL1 KD mutation testing is to help exclude the TKI(s) unlikely to be effective. A handful of mutations have been robustly associated with resistance to 2G and 3G TKIs: these are listed in Table 3. What about the others? For many (yet not for all) mutations, IC50 data are available and have been used to generate multicolored tables using a traffic-light code to indicate whether the mutation is expected to be sensitive, moderately resistant, or highly resistant to a given TKI.9,10,26 However, different tables sometimes report conflicting data—which highlights how different experimental conditions impact the raw IC50 data obtained. Therefore, although it is tempting to use a pocket version of one of these tables as a sort of “at-a-glance” guide to the TKI choice, caution should be exerted: measuring in vitro antiproliferative activity is an artificial way to rank inhibitors for their antileukemic activity in vivo in a much more complex system. Thus, for mutations other than those listed in Table 3, it is wiser to let clinical considerations regarding comorbidities, risk factors, and therapeutic endpoints specific to the patient prevail.
Mutations impacting the selection of the subsequent-line 2G TKI or of ponatinib
| Mutation | Contraindicated TKI(s) |
|---|---|
| Y253H | Nilotinib |
| E255K | Nilotinib, bosutinib |
| E255V | Nilotinib |
| V299L | Dasatinib, bosutinib |
| T315I | Dasatinib, nilotinib, bosutinib |
| T315A | Dasatinib |
| T315L | Ponatinib (asciminib?) |
| T315M | Ponatinib (asciminib?) |
| F317L | Dasatinib |
| F317V | Dasatinib |
| F317I | Dasatinib |
| F317C | Dasatinib |
| F359V | Nilotinib (asciminib) |
| F359I | Nilotinib (asciminib) |
| F359C | Nilotinib (asciminib) |
| Mutation | Contraindicated TKI(s) |
|---|---|
| Y253H | Nilotinib |
| E255K | Nilotinib, bosutinib |
| E255V | Nilotinib |
| V299L | Dasatinib, bosutinib |
| T315I | Dasatinib, nilotinib, bosutinib |
| T315A | Dasatinib |
| T315L | Ponatinib (asciminib?) |
| T315M | Ponatinib (asciminib?) |
| F317L | Dasatinib |
| F317V | Dasatinib |
| F317I | Dasatinib |
| F317C | Dasatinib |
| F359V | Nilotinib (asciminib) |
| F359I | Nilotinib (asciminib) |
| F359C | Nilotinib (asciminib) |
List of single mutations that, when detected, should exclude one or more 2G TKIs or ponatinib from the decision algorithm. All mutations except V299L, T315A, T315L, and T315M are also resistant to imatinib. Asciminib has been included, in brackets, for some mutations based on in vitro or in vivo data. This list will grow once asciminib resistance mutations are fully elucidated.
In case of warning, a gray area where continuation or change are equally allowed, detecting resistance mutations tilts the balance toward a change of therapy. But what does resistance mutation mean? From a clinical standpoint, interpreting BCR::ABL1 KD mutation testing results may not always be straightforward. What about mutations for which there are no IC50 data whatsoever? There is not necessarily a biunivocal link between resistance and mutations: some mutations might just be innocent bystanders. Comprehensive databases of mutations detected in TKI-resistant patients have never been built, so for less frequently occurring mutations, physicians often need to perform cumbersome literature searches for whether the mutation has ever been reported in any published study. And what about mutations detected at low levels, eg, by NGS? In case of warning, there is usually not a compelling urgency to intervene: when the relation between the mutation detected and the inadequate response is uncertain, it may be wise to repeat testing in 1 to 3 months. True resistance mutations are expected to expand, or at least persist, if therapy is not changed. Anyway, mutations are likely to be a gauge of the degree of genetic instability: thus, regardless of their actual role in resistance, positivity for any mutation identifies higher-risk patients requiring more careful monitoring.27
In general, when the clinical relevance of a detected mutation is uncertain (irrespective of whether this is due to the lack of sensitivity data or to a low mutational burden), closer monitoring of BCR::ABL1 transcript kinetics may help in deciding whether therapy should be switched.
Take-home message from the clinical case
In the case presented above, using NGS could have been beneficial. The E255K is well known to confer resistance to both imatinib and nilotinib. Thus, detecting a low-level E255K mutation would have prevented the unfortunate choice of a TKI (nilotinib) not active against this mutation. The case also exemplifies that if a mutant clone is not rapidly cleared, and if inefficiently inhibited BCR::ABL1 persists at high levels (as mirrored by the high levels of transcript), further acquisition of mutations, giving rise to CMs, may occur.
Conflict-of-interest disclosure
Simona Soverini has received speaker fees from Incyte Biosciences and Novartis.
Off-label drug use
Simona Soverini: There is nothing to disclose.
