

Abstract
Atypical chronic myeloid leukemia (aCML) is included in the group of myelodysplastic/myeloproliferative neoplasms by the International Consensus Classification and has been renamed as MDS/MPN with neutrophilia by the fifth edition of World Health Organization classification. It is always characterized by morphologic identification of granulocytic dysplasia with >10% circulating immature myeloid cells, 2 distinguished features that differentiate this disease among the others. Somatic mutations may help to diagnose but are not specifically pathognomonic of the disease, with the most detected including ASXL1, SETBP1, NRAS, KRAS, SRSF2, and TET2 and with low-frequency CBL, CSF3R, JAK2, and ETNK1. The genomic landscape of aCML has been recently unravelling, revealing that SETBP1 and ETNK1 are usually not ancestral but secondary events associated with disease progression. Unfortunately, until now, no consensus on risk stratification and treatment has been developed: Mayo Clinic prognostic score identified as adverse events age >67 years, hemoglobin level <10 g/dL, and TET2 mutations. Although some possible genetic markers have been identified, allogeneic transplant remains the only curative strategy.
Learning Objectives
Evaluate the diagnostic algorithm
Characterize the somatic mutations landscape
Analyze prognostic features
Summarize possible treatments
Introduction
Atypical chronic myeloid leukemia (aCML) is a clonal hematopoietic disease previously identified as a neoplasm with mixed dysplastic/myeloproliferative (MDS/ MPN) features in the absence of monocytosis and eosinophilia.1 It was introduced in the fourth edition of the World Health Organization classification in the subgroup of mixed MDS/MPN together with other entities, such as juvenile myelomonocytic leukemia, MDS/MPN with ring sideroblasts and thrombocytosis, and MDS/MPN unclassifiable.2 In the recent 2022 World Health Organization revision, aCML was retained as terminology with a clear distinction between this disorder and myeloid neoplasms with monocytosis and eosinophilia.3 The 2022 International Consensus Classification replaces the term aCML with MDS/MPN with neutrophilia, avoiding the possible confusion with typical chronic myeloid leukemia (Ph+ CML).4
CLINICAL CASE
A 62-year-old patient was admitted to the hospital for intense asthenia and widespread pain. Complete blood count showed mild anemia (10.7 g/dL), hyperleukocytosis (155 × 109/L), and absolute neutrophil count of 136 × 109/L. Splenomegaly was noted on physical examination (4 cm below costal margin), and peripheral blood smear showed neutrophilia (64% with >20% dysplastic features), in the absence of monocytosis. The absence of an abnormal karyotype on cytogenetic analysis, as well as the absence of BCR::ABL1 and other rearrangements in MPN driver genes, allowed to exclude the diagnosis of CML or Ph-negative MPNs. Bone marrow analysis showed dysplastic and markedly expanded granulopoiesis (myelo-erythroid ratio 9:1) and 3% of CD34+ blast cells, consistent with the morphologic suspect of aCML.
Clinical and morphologic features
aCML is a rare disease with an incidence of 1% to 2%; it affects elderly patients with a median age ranging between 60 and 70 years, with a male predominance.5-7 The disease is associated with a poor outcome, with a median overall survival (OS) of 10 to 28 months8 and a high rate of leukemic transformation (>15%–20% at 5 years).9 It is always characterized by leukocytosis with white blood cell count >13 × 109/L with dysplastic features in neutrophils and their precursor for >10% of the whole leukocyte population10 (Table 1). Dysplasia includes abnormal chromatin clumping, hypersegmented or Pelger-Huet forms, and cytoplasmatic hypogranularity.10 Monocytes must be <10% of total leukocytes, and the new International Consensus Classification includes also cytopenia defined as in MDS by anemia (hemoglobin level <13 g/dL in males and 12 g/dL in females), neutropenia with absolute neutrophil count <1.8 × 109/L, and thrombocytopenia with a platelet count <150 × 109/L.4 The upper limit of blast cells in all MDS/MPN entities is <20%. Bone marrow morphologic analysis showed hypercellularity, with a predominance of granulocytes showing marked dysplasia; more than half of patients showed also erythroid and some degree of megakaryocytic dysplasia. Variable degrees of increased reticulin fibrosis have been also reported.3,4,10 The differential diagnosis of aCML includes the following:
Diagnostic criteria for atypical chronic myeloid leukemia
| World Health Organization criteria | International Consensus Classification criteria |
|---|---|
| PB leukocytosis (WBC count ≥13 × 109/L) because of increased numbers of neutrophils and their precursors with prominent dysgranulopoiesis | Leukocytosis ≥13 × 109/L, due to increased numbers of neutrophils and their precursors (promyelocytes, myelocytes, and metamyelocytes), the latter constituting ≥10% of the leukocytes |
| Neutrophil precursors (promyelocytes, myelocytes, metamyelocytes) ≥10% of leukocytes | Dysgranulopoiesis, including the presence of abnormal hyposegmented and/or hypersegmented neutrophils ± abnormal chromatin clumping |
| Cytopenia (anemia, hemoglobin <13 g/dL in males, <12 g/dL in females; neutropenia, absolute neutrophil count <1.8 × 109/L; thrombocytopenia, platelets <150 × 109/L) | |
| Less than 20% blasts in the PB and BM | Blasts <20% of the cells in PB and BM |
| No or minimal absolute monocytosis; monocytes usually <10% of leukocytes | No or minimal absolute monocytosis; monocytes constitute <10% of the PB leukocytes |
| Minimal absolute basophilia; basophils usually <2% of leukocytes | No eosinophilia; eosinophils constitute <10% of the PB leukocytes |
| Hypercellular BM with granulocytic proliferation and granulocytic dysplasia, with or without dysplasia in the erythroid and megakaryocytic lineages | Hypercellular BM with granulocytic proliferation and granulocytic dysplasia, with or without dysplasia in the erythroid and megakaryocytic lineages |
| No Ph chromosome or BCR::ABL1 fusion gene and not meeting criteria for PV, ET, or PMF | No BCR::ABL1 or genetic abnormalities of M/L-Eo with TK gene fusions. The absence of MPN-associated driver mutations and the presence of SETBP1 mutations in association with ASXL1 provide additional support for a diagnosis of aCML. |
| No evidence of PDGFRA, PDGFRB, FGFR1 rearrangement, or PCM1::JAK2 |
| World Health Organization criteria | International Consensus Classification criteria |
|---|---|
| PB leukocytosis (WBC count ≥13 × 109/L) because of increased numbers of neutrophils and their precursors with prominent dysgranulopoiesis | Leukocytosis ≥13 × 109/L, due to increased numbers of neutrophils and their precursors (promyelocytes, myelocytes, and metamyelocytes), the latter constituting ≥10% of the leukocytes |
| Neutrophil precursors (promyelocytes, myelocytes, metamyelocytes) ≥10% of leukocytes | Dysgranulopoiesis, including the presence of abnormal hyposegmented and/or hypersegmented neutrophils ± abnormal chromatin clumping |
| Cytopenia (anemia, hemoglobin <13 g/dL in males, <12 g/dL in females; neutropenia, absolute neutrophil count <1.8 × 109/L; thrombocytopenia, platelets <150 × 109/L) | |
| Less than 20% blasts in the PB and BM | Blasts <20% of the cells in PB and BM |
| No or minimal absolute monocytosis; monocytes usually <10% of leukocytes | No or minimal absolute monocytosis; monocytes constitute <10% of the PB leukocytes |
| Minimal absolute basophilia; basophils usually <2% of leukocytes | No eosinophilia; eosinophils constitute <10% of the PB leukocytes |
| Hypercellular BM with granulocytic proliferation and granulocytic dysplasia, with or without dysplasia in the erythroid and megakaryocytic lineages | Hypercellular BM with granulocytic proliferation and granulocytic dysplasia, with or without dysplasia in the erythroid and megakaryocytic lineages |
| No Ph chromosome or BCR::ABL1 fusion gene and not meeting criteria for PV, ET, or PMF | No BCR::ABL1 or genetic abnormalities of M/L-Eo with TK gene fusions. The absence of MPN-associated driver mutations and the presence of SETBP1 mutations in association with ASXL1 provide additional support for a diagnosis of aCML. |
| No evidence of PDGFRA, PDGFRB, FGFR1 rearrangement, or PCM1::JAK2 |
BM, bone marrow; ET, essential thrombocythemia; M/L-Eo, myeloid/lymphoid neoplasms with eosinophilia; PB, peripheral blood; PMF, primary myelofibrosis; PV, polycythemia vera; TK, tyrosine kinase; WBC, white blood cell.
BCR1/ABL-positive CML, distinguished by not only the absence of specific t(9;22) but also the presence of dysgranulopoiesis and the almost normal basophil count (<2%) in aCML.
Chronic neutrophilic leukemia (CNL), including the presence of >10% of immature myeloid precursors and dysplasia, which are unique distinctive features of aCML. Mutational features may allow distinction with the CSFR3 mutation most frequently observed in CNL but not restricted only to this disease.
Chronic myelomonocytic leukemia (CMML), with the increased monocyte count exceeding more than 10% of the leukocyte count.
Prefibrotic myelofibrosis, in which the availability of myeloproliferative gene markers (JAK2, CALR, and MPL) may allow the possible distinction between the 2 forms. More difficult is the distinction in triple-negative myelofibrosis3,4 (Figure 1).
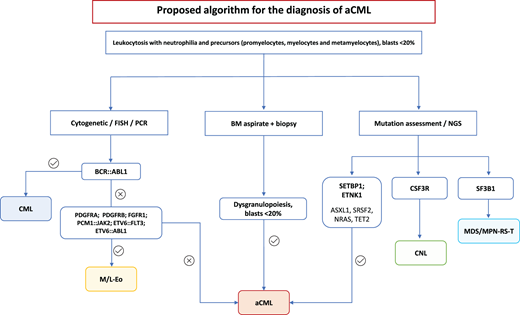
Algorithm proposed for the diagnosis of aCML. FISH, fluorescence in situ hybridization; MDS/MPN-RS-T, myelodysplastic/ myeloproliferative neoplasms with ring sideroblasts and thrombocytosis; M/L-Eo, myeloid/lymphoid neoplasms with eosinophilia; NGS, next-generation sequencing; PCR, polymerase chain reaction.
Algorithm proposed for the diagnosis of aCML. FISH, fluorescence in situ hybridization; MDS/MPN-RS-T, myelodysplastic/ myeloproliferative neoplasms with ring sideroblasts and thrombocytosis; M/L-Eo, myeloid/lymphoid neoplasms with eosinophilia; NGS, next-generation sequencing; PCR, polymerase chain reaction.
Genetic features
The reported frequency of chromosomal abnormalities in aCML is widely variable, ranging from 20% to 88% in different reports.5,6,11,12 The most common abnormalities reported were trisomy 8 or 9, del 20(q), −7/7(q), and isochromosomes 17q. Less frequently, aberrations in chromosomes 12, 13, 14, 19, and 21 have been described.5,6,11,12 Mutations are usually detected in all aCML cases: higher frequency for ASXL1, SETBP1, NRAS, KRAS, SRSF2, and TET2 has been reported, whereas CBL, CSF3R, JAK2, and ETNK1 were revealed with low frequency (<10% of cases).13 SETBP1 may support aCML diagnosis: identified in one-fourth of patients, it can be also found in MDS/MPN unclassifiable patients, in some CMML, occasionally in juvenile CMML, and in some secondary acute myeloid leukemia arising from MDS/MPN.14,15 SETBP1 can be associated with specific baseline features such as higher leukocyte count, low platelet count, and a worse prognosis.14-16 The mutation maps on chromosome 18q21.1 and encodes for SET binding protein, a negative regulator of the tumor suppressor protein phosphatase 2A (PP2A) with increased repression of activity and cellular proliferation.17,18 SETBP1 interacts with SET, protecting it from cleavage and allowing the creation of a complex (SETBP1/SET/PP2A) that increases proliferation and expansion of the leukemic clone.19-26 Piazza and colleagues14,27 clearly demonstrated that most SETBP1 somatic mutations cluster in a mutational hotspot within the SKI-homologous region of the protein, conferring a proliferative advantage to the mutated cells and protecting the mutation from ubiquitination. Most SETBP1 mutations are located within a 14-amino-acid stretch (codons 858-871), which is also mutated in Schinzel-Giedion syndrome, a rare genetic disease characterized by congenital malformations, mental retardation, and frequent epithelial tumors.
SETBP1 mutations have shown a strong association with ASXL1 and CBL mutations and are mutually exclusive of JAK2 and TET2 mutations.14,28 SETBP1 also has a complex role as a transcriptional modulator, considering its ability in direct interaction with genomic DNA (recruitment of HCF1, KMT2A, PHF8, PHF6), the possible binding capacity to MDS1 and EVI1 complex locus protein EVI1 or MECOM (upregulation of several genes involved in stem cell proliferation and myeloid differentiation), self-renewal of myeloid progenitors, and RUNX1 downregulation.27,29 A study conducted on 71 patients showed that most were ASXL1 positive (92%), and SETBP1 was detected in 38%. Clonal hierarchy was identified, and ASXL1 was acquired early, whereas SETBP1 was never reported as being ancestral but always secondary in disease progression11 (Figure 2).
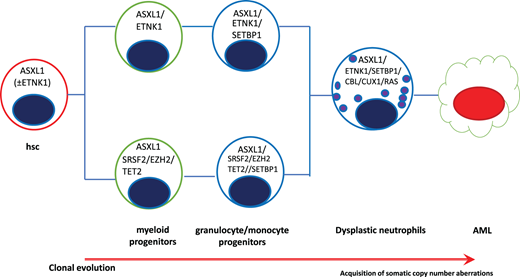
Clonal hierarchy in aCML showing the progressive acquisition of somatic mutations. AML, acute myeloid leukemia.
Clonal hierarchy in aCML showing the progressive acquisition of somatic mutations. AML, acute myeloid leukemia.
Recurrent mutations in ETNK1 have emerged as being relatively specific for aCML (up to 9% of cases) and CMML (3% of cases) and not found in other myeloid diseases.30 The ETNK1 gene maps on chromosome 12p12.1 and encodes a protein known also as ethanolamine kinase, which acts as catalyzer for the biosynthesis of phosphatidylethanolamine through the Kennedy pathway, responsible for the de novo synthesis of membrane phospholipids.31,32 These mutations cluster in a small region of the kinase domain, encoding for H243Y and N244S (1/8 H243Y; 7/8 N244S) and G245 V/A, all as heterozygous and present in the dominant clone.33 ETNK1 mutations decrease the activity of the enzyme, reducing the synthesis of phosphoethanolamine, increasing the mitochondrial activity with final increased reactive oxygen species production, DNA damage, and genomic instability.34,35 On the hierarchical scale, it seems that ETNK1 mutation may precede ASXL1 and SETBP1.34,35
In summary, studies on the clonal architecture of aCML showed that ETNK1 and ASXL1 are ancestral mutations, with RAS, CBL, TET2, SRSF2, and SETBP1 as secondary events; CBL mutations have a tendency to reach homozygosity through somatic uniparental disomy.10 CCND2 mutations also have been recently suggested in patients with aCML. Some groups36,37 reported CCND2 mutations at low variant allelic frequencies: Khanna et al36 showed, in a cohort of 116 patients with Ph-negative MPN, CCND2 mutations concomitant in 1 case to SETBP1 and in 1 case to SRSF2 mutation. CCND2 mutation in the P281 codon resulted in the accumulation of degradation-resistant cyclin in D2, with predominant staining in nuclear localization regardless of the cycle cell phase. This peculiar localization seems to be responsible for prolonged cell survival without conferring growth factor independence.38 Recently, Carreño-Tarragona and colleagues39 compared the clinical and genomic profile of aCML and CNL: they suggested that apart from CSFR3 more commonly mutated in CNL, as well as EZH2 and TET2 more commonly mutated in aCML, no differences were found in the pathways affected, suggesting that CNL and aCML are a continuum of the same disease.
Different groups analyzed gene expression profiling in aCML: a significant change in the expression levels of SETBP1, CDKN2A, GATA2, MPL, TMEM14C, CSF3R, and FLT3 genes was observed in a group of 26 patients compared to 59 patients with CMML.40 Indeed, Zhang and colleagues41 reported 3 different clusters in 158 samples of different myeloid disorders, including CNL, aCML, MDS/MPN unclassifiable, MDS/MPN, and CMML, showing dissimilarity of different combinations of mutant patterns without specific clusterization within any one diagnosis. Fontana et al42 showed 2 different aCML groups based on gene expression with overexpression of 3 different genes (PARP1, DNPH1, and GFI1B) associated with a worse prognosis.
CLINICAL CASE (continued)
Next-generation sequencing was performed, and mutations were identified in ASXL1 (29% VAF), CBL (4% variant allele frequency), SETBP1 (44% VAF), and SRSF2 (43% VAF), with a compound mutation in CCND2 (c.841C>T, 28% VAF and c.842C>G, 4% VAF). In this case, in consideration of the mutational profile detected on next-generation sequencing, an indication for allogenic transplant has been made and human leukocyte antigen typing is being carried out, with a search for possible bone marrow stem cell donors.
Risk stratification and prognosis
Considering the rarity of the disease, no consensus on a possible risk stratification has been reported. Two studies showed that age >65 years, female sex, hemoglobin <10 g/dL, leukocyte count >50 × 109/L, and immature circulating precursors were adverse prognostic factors.6,9 Wang et al7 reported that leukocytosis and immature myeloid cells but not the dysgranulopoiesis maintained the negative role associated with shorter OS in a large series of patients, including 65 patients with aCML with a median OS of 12.4 months.
In 2017, Mayo Clinic developed a model based on a small data set of 25 patients. In this proposed model, advanced age, low hemoglobin, and TET2 mutations were considered of significance in multivariate analysis. Two categories of patients were identified (low, with 0-1 risk factor, and high risk with ≥2 risk factors) considering 1 point each for age >67 years, hemoglobin level <10 g/dL, and detection of the TET2 mutation. The median OS observed was 18 months for the low-risk category and 7 months for the high-risk group.5 Palomo et al11 explored the effects of mutations as prognostic indicators: while ASXL1 did not retain a prognostic impact considering that most patients were positive, RUNX1, CUX1, and NRAS were associated with a shorter OS and SRSF2 and SETBP1 with positive outcome.
Treatment
No standard of care exists for the treatment of aCML. In addition, no consensus recommendations or risk-based treatment algorithms exist to help guide a watch-and-wait approach vs initiation of therapy. The most common administered therapy is hydroxyurea (HU), typically used to control leukocytosis or symptomatic splenomegaly. There have been multiple reports of HU inducing complete and partial hematologic responses in aCML, but the duration of response is usually limited to a few months.6,9,28,43 Interferon α (IFNα) has also been associated with partial or sometimes complete hematologic response but also with discontinuation due to toxicity.28,43-45 A phase 2 study of pegylated-IFNα-2b was shown to have improved tolerability over standard IFNα in BCR-ABL1–negative MPNs.46 This long- acting formulation is associated with a better toxicity profile and offers a treatment option for those ineligible for clinical trials. Both drugs are usually used in a palliative setting, in the absence of a possible allogeneic transplant (hematopoietic stem cell transplant) procedure strategy.
Progressive anemia with development of transfusion dependence is common in aCML, contributing to increased morbidity. Splenectomy is generally not recommended in the management of this disease given its limited clinical response, anecdotal risk of accelerated neutrophilia, and relatively high perioperative morbidity already known in patients with MPN.43,44 Erythroid- stimulating agents also have limited data in aCML, with only 1 study reporting a poor response.47 Immunomodulating agents, such as thalidomide and lenalidomide, were also tested with scarce responses.28 Even if not considered a standard of care, experiences with hypomethylating agents (HMAs) were described: the Mayo Clinic reported a limited experience in 5 patients with a 40% rate of stable disease.5 Decitabine was reported in several other anecdotical cases with a complete remission rate after almost 4 cycles.48-51
Rarely, CSF3R mutation can be detected in aCML with constitutive activation of JAK-STAT signaling due to T615A, T618I, and T640N mutations. The potential benefit of ruxolitinib in CSF3R T618I–mutated disease was first demonstrated in a patient with CNL with CSF3R T618I who achieved a marked reduction in neutrophilic leukocytosis and improvement of anemia and thrombocytopenia.52 Subsequently, a patient with hydroxyurea-refractory aCML treated with ruxolitinib escalated from 10 to 20 mg twice daily resulted in similar hematologic improvements (reduced splenomegaly and peripheral blood myeloid immaturity, reverted weight loss, and improved symptom scores, without a change in CSF3R mutant allele frequency).53 A phase 2 trial enrolled 44 patients (23 with aCML and 21 with CNL). Of the aCML cohort, 6 patients had a CSFR3 mutation: only 2 patients obtained a partial response, and grade 3 anemia and thrombocytopenia were observed in 34% and 14% of patients, respectively.54
Allogeneic transplant remains the only potential curative strategy: a large series of 42 patients was described by Onida et al.55 Sixty-four percent of patients underwent a matched sibling donor transplant, with reduced conditioning in 24% of them. The 5-year relapse-free survival was 36%, transplant-related mortality was 24%, and the relapse rate was 40%. Age and European Society for Blood and Marrow Transplantation score were the identified independent prognostic factors for OS. Contrasting results were reported by other small studies: Koldehoff et al56 described favorable outcomes with over 80% OS at 5 years in 21 patients who had received allogeneic HSCT, whereas significantly worse outcomes were described by Mittal et al57 due to high transplant-related mortality from graft-vs-host disease and sepsis.
Other actionable targets have been identified and small- molecule inhibitors tested: trametinib, a MEK1/2 inhibitor, inhibits the extracellular signal–regulated kinase directly downstream from the mitogen-activated protein kinase pathway. A single patient with aCML harboring a NRAS mutation attained a near-complete hematologic response with 14 months of disease control with trametinib.42 Another patient obtained a 3-month improvement of leukocytosis and splenomegaly.58
Dasatinib, a dual inhibitor of BCR::ABL1 and SRC family kinase, could have potential therapeutic value in aCML: in vitro studies of cell lines with CSF3R-truncated mutations have demonstrated dysregulation in the SRC family–TNK2 kinases with sensitivity to dasatinib.52 No in vivo reports have been published so far. Venetoclax also could be a possible option in the future associated with HMA in this setting.59 The ABNL-MARRO (A Basket Study of Novel Therapy for Untreated MDS/MPN and Relapsed/Refractory Overlap Syndromes) is an ongoing recruiting phase 1/2 study that tests novel treatment combinations in MDS/MPNs. The exploratory objectives of the study include investigating genetic biomarkers of response and characterizing molecular responses in this setting of patients. The first open arm includes the JAK1 inhibitor itacitinib, as well as oral decitabine plus cedazuridine (ASTX727).60
Conclusions
aCML is a rare myelodysplastic/myeloproliferative neoplasm characterized by increasing leukemic cell burden, organomegaly, anemia, and bone marrow failure. Some negative prognostic factors associated with a shorter survival have been identified, such as age >65 years, female sex, leukocytosis >50 × 109/L, and SETBP1 mutations. The molecular pathogenesis of aCML is heterogeneous, with mutations involving SETBP1, CSF3R, ASXL1, and ETNK1 the most studied to date. The clonal hierarchy in the mutational landscape has been recently proposed.
Unfortunately, no consensus on treatment has been reported until now, and allogeneic transplant remains the only valid option. Palliative chemotherapy (HU, IFNα) is the most common treatment in patients not eligible for the transplant procedure. HMAs can be a bridge to the transplant approach, but the results reported, based on small cohort of patients, are not encouraging. Targeted therapies with JAK2 inhibitor (ruxolitinib), SRC kinase inhibitor (dasatinib), and MEK inhibitor (trametinib) in patients with aCML with actionable target mutations have shown some interesting responses, but large cohorts of patients are needed to understand if they could represent a future therapeutic strategy.
Conflict-of-interest disclosure
Massimo Breccia received honoraria by Novartis, Incyte, Pfizer, BMS, Abbvie, AOP, and Jazz.
Off-label drug use
Massimo Breccia: All of the new actionable target treatments are off-label.
