

Abstract
Progression to myelodysplastic syndromes (MDS) and acute myeloid leukemia is one of the most serious complications of the inherited bone marrow failure and MDS-predisposition syndromes. Given the lack of predictive markers, this risk can also be a source of great uncertainty and anxiety to patients and their providers alike. Recent data show that some acquired mutations may provide a window into this risk. While maladaptive mechanisms, such as monosomy 7, are associated with a high risk of leukemogenesis, mutations that offset the inherited defect (known as somatic genetic rescue) may attenuate this risk. Somatic mutations that are shared with age-acquired clonal hematopoiesis mutations also show syndrome-specific patterns that may provide additional data as to disease risk. This review focuses on recent progress in this area with an emphasis on the biological underpinnings and interpretation of these patterns for patient care decisions.
Learning Objectives
Understand the types of somatic clonal mutations in inherited bone marrow failure/MDS syndromes
Identify somatic genetic rescue mutations in specific bone marrow failure/MDS syndromes
Understand how to diagnosis rescue mutations in clinical practice
Introduction
The stressed hematopoietic system in individuals with inherited bone marrow failure and myelodysplastic syndrome (MDS) predisposition syndromes is remarkably adaptable. Due to the high turnover nature of hematopoiesis, de novo adaptations that provide a fitness advantage, either in terms of survival or proliferation, compete with the germline failing hematopoiesis and persist over time.1,2 These adaptive mechanisms are remarkably varied, with some exquisitely specific for a single inherited disorder while others are shared across different syndromes broadly. Here, I will discuss these mechanisms of adaptation and examine how these findings may impact risk stratification and inform patient care. I will review general progress in this area, but for illustrative purposes and because of the predominance of the recent evidence, I will focus on three disorders: (1) SAMD9/9L syndromes, a common cause of monosomy 7 MDS in early childhood; (2) Shwachman- Diamond syndrome, a ribosomopathy associated with MDS and acute myeloid leukemia (AML) in later childhood and young adults; and (3) the short telomere syndromes, a common adult-onset inherited syndrome.
Diagnosis of genetic predisposition to MDS/AML informs patient care
Syndromes associated with genetic predisposition to MDS and/or AML include both the inherited bone marrow failure (BMF) syndromes and MDS predisposition syndromes; these will collectively be referred to as BMF/MDS syndromes in this review.3 These genetic disorders are diverse in their etiology but share a risk of myeloid neoplasm evolution as a hallmark with variable risk of antecedent marrow failure, extra-hematopoietic manifestations, and non-myeloid cancers. At least 14 common syndromes and roughly 100 genes have been linked to BMF/MDS syndromes due to defects in in many mechanisms, including DNA repair, ribosome function, telomere maintenance, or hematopoietic transcription factor signaling, among others.4,5 The mechanism for more recently identified syndromes, such as the RNA helicase DDX41 and SAMD9/9L, whose gene products suppress hematopoiesis, remains poorly understood.6-8 Some syndromes present with severe cytopenia or immunodeficiency in early childhood, and others can remain cryptic until adulthood.9 Even within a single genetic disorder, there can be variable severity and heterogeneity in presentation as well as variable risk of leukemogenesis. Distinguishing these presentations from de novo disease is clinically relevant because of implications for treatment, long-term surveillance, and family counseling.10,11 While some BMF/MDS syndromes present with progressive stem cell failure, in a number of cases, the first presentation may be as an MDS or overt AML.12 Timely diagnosis of an underlying genetic predisposition has critical implications for timing of hematopoietic stem cell transplant, related donor selection, preparative regimens, and posttransplant care.13,14
Age-dependent presentation of MDS/AML in inherited BMF/MDS syndromes
The onset of MDS/AML in these syndromes has some age- dependent patterns with SAMD9/9L syndromes and GATA2 deficiency presenting more often in childhood.15,16 In older adults, mutations that cause telomere shortening (collectively known as the short telomere syndromes17 ) and germline DDX41 mutations account for the majority of known inherited predisposition.6,18 RUNX1-familial platelet disorder with associated myeloid malignancies (FPDMM) is more rare and can present at nearly any age with myeloid or lymphoid malignancy.19,20 Other more rare autosomal recessive inherited causes of MDS/AML that are seen in both children and younger adults include Shwachman-Diamond syndrome and Fanconi anemia.21,22 Figure 1 summarizes the age-dependent patterns of MDS/AML diagnosis in these frequently diagnosed syndromes and reflects the peaks and ranges of MDS/AML diagnosis obtained from numerous published family and cohort studies.
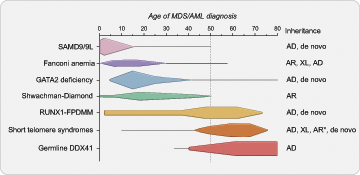
Age-dependent presentation of myeloid malignancy in inherited bone marrow failure and MDS predisposition syndromes. A schematic demonstrating the typical and peak ages of myeloid malignancy diagnosis in individuals with inherited bone marrow failure and MDS predisposition syndromes. There is an age-dependent onset depending on the underlying genetic cause that informs approaches to surveillance and patient counseling. Schema is based on a comprehensive review of myelodysplastic syndrome (MDS) and/or acute myeloid leukemia (AML) cases reported in the literature in individuals with confirmed germline predisposition. The reported incidence of MDS/AML in these disorders is impacted by rates of hematopoietic stem cell transplant for bone marrow failure and other competing risks, such as pulmonary fibrosis in the short telomere syndromes which accounts for the drop off of MDS/AML diagnosed in older ages. Horizontal lines demonstrate the published age range where there are isolated cases at those age extremes. Queried publications are referenced in reference 13 in addition to references 23, 51, and 72 and PubMed IDs 32088370, 30578959, and 34469508. *Autosomal recessive inheritance is rarely found in the short telomere syndromes (<5%) and more often manifests as aplastic anemia with few reports of MDS/AML in these individuals. AD, autosomal dominant; AR, autosomal recessive; XL, X-linked.
Age-dependent presentation of myeloid malignancy in inherited bone marrow failure and MDS predisposition syndromes. A schematic demonstrating the typical and peak ages of myeloid malignancy diagnosis in individuals with inherited bone marrow failure and MDS predisposition syndromes. There is an age-dependent onset depending on the underlying genetic cause that informs approaches to surveillance and patient counseling. Schema is based on a comprehensive review of myelodysplastic syndrome (MDS) and/or acute myeloid leukemia (AML) cases reported in the literature in individuals with confirmed germline predisposition. The reported incidence of MDS/AML in these disorders is impacted by rates of hematopoietic stem cell transplant for bone marrow failure and other competing risks, such as pulmonary fibrosis in the short telomere syndromes which accounts for the drop off of MDS/AML diagnosed in older ages. Horizontal lines demonstrate the published age range where there are isolated cases at those age extremes. Queried publications are referenced in reference 13 in addition to references 23, 51, and 72 and PubMed IDs 32088370, 30578959, and 34469508. *Autosomal recessive inheritance is rarely found in the short telomere syndromes (<5%) and more often manifests as aplastic anemia with few reports of MDS/AML in these individuals. AD, autosomal dominant; AR, autosomal recessive; XL, X-linked.
Common inherited causes of childhood-onset MDS
Among young children with MDS, germline mutations in SAMD9 or SAMD9L are the most common inherited cause. Mutations in these genes account for 8%-17% of pediatric MDS and up to 40% in children with monosomy 7 MDS who are younger than 5 years.16,23 SAMD9 and SAMD9L are homologous genes located on chromosome 7q21; they have proposed roles in antiviral immunity and protein translation and also function to negatively regulate cellular proliferation.8,24 While their precise function is not known, in experiments of hematopoietic cells in culture, expression of pathogenic germline SAMD9 or SAMD9L mutations enhances the growth suppressing effect of the wild-type protein, resulting in a profound inhibition of cell growth. As such, mutations in these genes mediate their effect through gain-of-function.8,25
In older children and adolescents, mutations in GATA2 predominate, accounting for approximately 10% of cases of pediatric MDS and up to 50% of monosomy 7 MDS in children who are older than 12 years.23,26 GATA2 is a zinc finger transcription factor that is required for maintenance and proliferation of hematopoietic and immune cells.27 Germline coding and noncoding mutations in GATA2 cause loss of function of the mutated allele and exert their effect through haploinsufficiency.28,29
Germline predisposition to MDS/AML in older adults
At the other end of the age spectrum, among adults over age 50 with MDS/AML, the short telomere syndromes are among the most common inherited causes.18,30-32 Mutations in genes encoding telomerase components and the other telomere maintenance genes, which are associated with telomere shortening, explain 5%-10% of suspected, but clinically unresolved, BMF/MDS syndromes in children and young adults.33,34 Among unselected MDS in adults, mutations in the telomerase reverse transcriptase (TERT) gene alone account for 3% of cases.32 This supports an even higher prevalence of the short telomere syndromes in adult-onset MDS, as germline loss-of-function mutations in TERT, resulting in TERT haploinsufficiency, account for the genetic cause in only half the cases.30 The genetic basis and mechanisms of telomere shortening for the other 15 germline mutant genes have been recently reviewed.9 In the bone marrow, short telomeres result in slowly progressive hematopoietic stem cell loss that leads to both BMF and primary immunodeficiency.35,36 Telomere length is the primary determinant of disease severity in the short telomere syndromes. Among children with more severe forms of telomere shortening, stem cell depletion usually manifests as aplastic anemia. In contrast, among older adults with relatively milder telomere shortening deficits, the risk of MDS/AML is higher and up to 15% for those over age 50.18,37 However, even among those over age 50 with MDS/AML, two-thirds of the patients suffer from extrahematopoietic disease, and the primary cause of death is pulmonary disease.18,38
MDS/AML comprise nearly 70% of all short telomere syndrome malignancies, with the remaining cancers being mostly squamous cancers that arise in the setting of a T-cell immunodeficiency.30,36 Although genome instability has been the primary hypothesized mechanism of carcinogenesis, short telomere syndromes have a lower than expected incidence of most cancers, and somatic alterations that arise in the setting of BMF, rather than genome instability, are the primary drivers of MDS/AML progression. The somatic landscape of the rare solid tumors seen in these patients is also quiescent and lacks the hallmarks of genome instability consistent with T-cell surveillance defects being their primary driver. Of note, a recently recognized entity associated with abnormally long telomere length has also been linked to the risk of myeloid as well as lymphoid neoplasms and clonal hematopoiesis. The risk of malignancy in this long telomere syndrome is associated with extended replicative potential of hematopoietic stem cells, and these disorders, in contrast to the short telomere syndromes, lead to hyperproliferative disease.9,39
In contrast to the short telomere syndromes, germline DDX41 mutations do not have obvious extrahematopoietic phenotypes and are more frequently are diagnosed in the setting of seemingly de novo MDS and AML.40 Germline mutation in DDX41 accounts for 2%-4% of unselected MDS/AML cases and represents a late-onset genetic predisposition, with a median age of MDS/AML diagnosis in the 7th decade.41,42 While initial reports were not associated with preceding bone marrow failure, recent data demonstrate antecedent cytopenia in nearly half of patients presenting with myeloid neoplasms due to germline DDX41 mutations.41 DDX41, located on chromosome 5q35, encodes an RNA helicase, and germline mutations have been proposed to alter pre-mRNA splicing and RNA processing.6 DDX41 is thought to have a tumor suppressor function whereby second-hit loss-of-function mutation or acquired del(5q) involving the wild-type DDX41 allele is seen at time of MDS/AML diagnosis in up to 60% of germline DDX41 cases.6
Distinct types of clonal hematopoiesis in inherited bone marrow failure/MDS predisposition syndromes
Clonal hematopoiesis (CH) is an overrepresentation of cells with an acquired somatic mutation, and this phenomenon is common in patients with inherited BMF/MDS syndromes. Hematopoietic stem cells harboring a mutation that results in a fitness advantage are selected and can rise to detectable levels. In some BMF/MDS syndromes, there are more selective pressures due to the underlying hypofunctioning bone marrow. In individuals with BMF/MDS syndromes, these acquired changes generally fall into two categories: (1) mutations that are commonly shared across many disorders and also seen with age-related clonal hematopoiesis (or clonal hematopoiesis of indeterminate potential, CHIP),43 and (2) those that are specific to one particular disorder.
CHIP in BMF/MDS syndromes
CH mutations that are age-related fall in a relatively small subset of genes, with approximately one dozen genes responsible for the vast majority of clonal hematopoiesis seen in unselected populations.43-45 High-risk somatic mutations in leukemia driver genes are the largest identifiable risk factor for hematologic malignancy in the general population.46 Several BMF/MDS syndromes demonstrate premature onset of age-related CH several decades earlier than the general population, suggesting antecedent CH may contribute to MDS/AML risk (Table 1).18,19,47,48 For example, TP53 mutations are common among some BMF/MDS syndromes. In the short telomere syndromes, somatic TP53 mutations allow the cell to bypass the short telomere checkpoint; these mutations are seen in approximately 15% of adults with short telomere syndromes but are distributed evenly between those with and those without MDS/AML.18,49 Among the TP53-mutant short telomere patients without MDS/AML, pulmonary fibrosis was the leading cause of mortality, not their hematologic disease. In children with Shwachman-Diamond syndrome, ultradeep sequencing identified somatic TP53 mutations in nearly half of those studied, although there was no association with hematologic phenotype.50 Consistent with sporadic MDS, acquisition of biallelic TP53 alterations through copy-neutral loss of heterozygosity (CN-LOH) at 17p, TP53 gene deletion, or second- hit mutation was associated with MDS/AML in both the short telomere syndromes and Shwachman-Diamond syndrome.47,49 In single-cell analysis of one individual with Shwachman-Diamond syndrome and p53-mutant AML, CN-LOH was documented subclinically (variant allele fraction [VAF] <0.1%) 4 years prior to AML diagnosis, suggesting that more advanced clinical diagnostic modalities may be able to identify the patients with exceptionally high risk of transformation to MDS/AML and allow early intervention.47 However, not all the inherited syndromes cause antecedent CHIP and, in individuals with germline mutations in DDX41, for example, progression to MDS/AML is instead associated with a second hit in the wild-type DDX41 allele (Table 2).51 Other recurrent CHIP mutations documented in inherited BMF/MDS syndromes have been recently reviewed.52
Risk of myeloid malignancy and age-related clonal hematopoiesis in inherited BMF/MDS syndromes
| Incidence of MDS/AML | Peak age of MDS/AML diagnosis | Prevalence of CHIP in carriers without hematologic malignancy | |
|---|---|---|---|
| SAMD9/9L syndromes | Moderate* | Early childhood | Absent |
| Fanconi anemia | 30%-40% | Childhood | ** |
| GATA2 deficiency | 75% | Late childhood through young adult | 20%-50% |
| Shwachman-Diamond syndrome | 10%-30% | Late childhood through young adult | 60% TP53 Other CHIP <10% |
| RUNX1-FPDMM | 20%-40% | Any age | 25%-60% |
| Short telomere syndromes | 15% | Adults >50 years | 30% |
| Germline DDX41 | 40%-50% | Adults >60 years | Rare |
| Incidence of MDS/AML | Peak age of MDS/AML diagnosis | Prevalence of CHIP in carriers without hematologic malignancy | |
|---|---|---|---|
| SAMD9/9L syndromes | Moderate* | Early childhood | Absent |
| Fanconi anemia | 30%-40% | Childhood | ** |
| GATA2 deficiency | 75% | Late childhood through young adult | 20%-50% |
| Shwachman-Diamond syndrome | 10%-30% | Late childhood through young adult | 60% TP53 Other CHIP <10% |
| RUNX1-FPDMM | 20%-40% | Any age | 25%-60% |
| Short telomere syndromes | 15% | Adults >50 years | 30% |
| Germline DDX41 | 40%-50% | Adults >60 years | Rare |
Lack of cohort studies limits definition of MDS/AML prevalence in SAMD9/9L syndromes.
Large-scale NGS studies in individuals with Fanconi anemia without MDS/AML have not been published.
RUNX1-FPDMM, RUNX1 familial platelet disorder with associated myeloid malignancies.
Common adaptive somatic genetic rescue mechanisms and maladaptive responses in inherited bone marrow failure and MDS predisposition syndromes and their association with risk of progression to MDS/AML
| Lower risk (adaptive rescue) | Higher risk (maladaptive response) | |
|---|---|---|
| SAMD9/9L syndromes | Second-site loss-of-function mutation (cis) UPD 7q | -7 / del7q |
| Shwachman-Diamond syndrome | EIF6 inactivating mutation Deletion 20q Isochromosome 7q | TP53 mutation |
| GATA2 deficiency | Direct reversion* | -7 / del7q / der(1;7) |
| Fanconi anemia | Direct reversion | |
| RUNX1-FPDMM | UPD 21q** | Somatic 2nd-hit RUNX1 mutation (trans) |
| Short telomere syndromes | Direct reversion TERT promoter mutation POT1 loss-of-function mutation RNA exosome mutation | -7 / del7q / der(1;7) TP53 mutation |
| DDX41 | — | Somatic 2nd-hit DDX41 mutation (trans) |
| Lower risk (adaptive rescue) | Higher risk (maladaptive response) | |
|---|---|---|
| SAMD9/9L syndromes | Second-site loss-of-function mutation (cis) UPD 7q | -7 / del7q |
| Shwachman-Diamond syndrome | EIF6 inactivating mutation Deletion 20q Isochromosome 7q | TP53 mutation |
| GATA2 deficiency | Direct reversion* | -7 / del7q / der(1;7) |
| Fanconi anemia | Direct reversion | |
| RUNX1-FPDMM | UPD 21q** | Somatic 2nd-hit RUNX1 mutation (trans) |
| Short telomere syndromes | Direct reversion TERT promoter mutation POT1 loss-of-function mutation RNA exosome mutation | -7 / del7q / der(1;7) TP53 mutation |
| DDX41 | — | Somatic 2nd-hit DDX41 mutation (trans) |
Direct reversion in Fanconi anemia and the short telomere syndromes encompasses multiple mechanisms, including uniparental isodisomy of the wild-type allele, somatic second-site loss-of-function mutation, and back mutation, that individually are seen more rarely.
Single case report by Catto et al found somatic reversion of a germline nonsense mutation in GATA2 to a synonymous mutation in an asymptomatic 61-year-old adult.54
Single case report by Glembotsky et al of somatic reversion via uniparental isodisomy of chromosome 21 in an individual with germline RUNX1 mutation and gradual improvement platelet number and function.75
RUNX1-FPDMM, RUNX1 familial platelet disorder with associated myeloid malignancies; UPD, uniparental isodisomy.
Somatic genetic rescue in BMF/MDS syndromes
Somatic genetic rescue (SGR) is a term that encompasses disease-specific genetic changes found in individuals with Mendelian hematopoietic and immunodeficiency disorders.53 Initially thought to be rare events, the availability of deep sequencing underscores that SGR is common and explains some of the varied penetrance of bone marrow failure and myeloid malignancy in BMF/MDS syndromes.25,49,54,55 Mechanisms of SGR can directly correct the mutant genotype (a process termed reversion) or can function indirectly to offset the effect. These mechanisms improve fitness on the cellular level, but, for the bone marrow as a whole, some mechanisms are beneficial (adaptive SGR) while others are pro-leukemogenic (maladaptive SGR). Understanding SGR mechanisms is important for patient care because adaptive mutations that theoretically offset the inherited defect and associated pressures toward leukemogenesis may identify a low-risk subset of patients.
The presence of beneficial versus leukemogenic adaptation influences disease course in SAMD9/9L syndromes
SGR has been documented in over half of individuals with SAMD9/9L syndromes and this occurs by varied mechanisms.23–25 One is the acquisition of a second SAMD9 or SAMD9L mutation in cis that offsets the effect of the germline mutation (termed second-site mutation). Another is the removal of the mutant allele through one of several processes, including (1) mitotic recombination, resulting in uniparental isodisomy of chromosome 7q (UPD7q), (2) aneuploidy, resulting in monosomy 7 or, more rarely, (3) focal gene deletion encompassing the mutant gene (Figure 2).23,25,55 In in vitro studies, coexpression of the second-site somatic loss-of-function mutation with the germline gain-of-function mutation ameliorated the growth suppressive defect.23,24 Interestingly, second-site somatic mutations can be either truncating or missense mutations, and both types show comparable restoration of growth in vitro.23 UPD7q, detected as CN-LOH at 7q by microarray, reverts cells harboring it to the wild-type genotype and is hypothesized to be a more complete rescue effect.23 In contrast, preferential loss of chromosome 7 or 7q containing the germline mutation, resulting in monosomy 7 or del(7q), is a maladaptive, pro-leukemogenic event that, as in other diseases, is associated with development of MDS/AML, most likely due to haploinsufficiency of additional genes on 7q.56 Unique to SAMD9/9L syndromes, there are reports of monosomy 7 clones disappearing and the patient obtaining hematologic and morphologic remission.23,25 Alternatively, and perhaps more often, the monosomy 7 clone acquires additional leukemia driver mutations in genes such as SETBP1, ASXL1, RUNX1, EZH2, and Ras pathway genes with high risk of disease progression.23
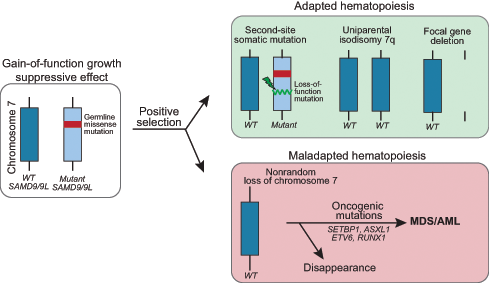
Mechanisms of somatic genetic rescue in the SAMD9/9L syndromes. Pathogenic germline mutations in SAMD9 and SAMD9L are gain-of-function and have a growth suppressive effect on hematopoiesis. Cells that acquire adaptations that improve growth or survival are selected in the high turnover environment of the bone marrow. Somatic second-site loss-of-function mutations in cis or removal of the mutant allele via uniparental isodisomy of chromosome 7q or focal gene deletion are not associated with progression to MDS/AML. In contrast, the monosomy 7 clone often acquires additional leukemia driver mutations with high risk of disease progression. WT, wild type.
Mechanisms of somatic genetic rescue in the SAMD9/9L syndromes. Pathogenic germline mutations in SAMD9 and SAMD9L are gain-of-function and have a growth suppressive effect on hematopoiesis. Cells that acquire adaptations that improve growth or survival are selected in the high turnover environment of the bone marrow. Somatic second-site loss-of-function mutations in cis or removal of the mutant allele via uniparental isodisomy of chromosome 7q or focal gene deletion are not associated with progression to MDS/AML. In contrast, the monosomy 7 clone often acquires additional leukemia driver mutations with high risk of disease progression. WT, wild type.
Multiple distinct SGR events can converge in one individual, and many different rescue mechanisms can be found in the same family (Figure 3A). Single-cell data recently confirmed these SGR events are mutually exclusive on the cellular level.23 In a cohort of children with MDS and SAMD9/9L syndromes, monosomy 7 predominated (55% with monosomy 7 at the time of MDS diagnosis).23 Of note, nearly half (18 of 37) of the children with monosomy 7 MDS also had concurrent somatic second-site mutations or UPD7q supporting monosomy 7 as a stronger driver of the hematologic phenotype toward MDS/AML than the adaptative mechanisms. However, family-based studies of children and adults with SAMD9/9L syndromes without MDS suggests adaptive UPD7q and somatic second-site mutations with remission potential may be more common than monosomy 7 overall, and there are many examples of carriers with sizable or multiple adaptive SGR clones experiencing a benign hematologic course into adulthood.25,55 Assessing for somatic second-site SAMD9/9L mutations, UPD7q, and monosomy 7 at initial SAMD9/9L syndrome diagnosis can be done through clinically available testing, and their detection, in conjunction with peripheral blood count monitoring, can improve risk stratification and tailor surveillance interventions.
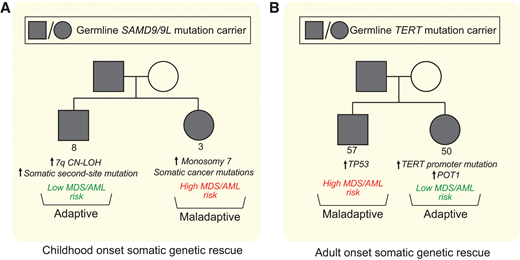
Distinct somatic genetic rescue mechanisms can be present within the same family and impact clinical course. Representative pedigrees of SAMD9/9L syndrome (A) and short telomere syndromes (B) showing that within 1 family carrying the same germline mutation, different somatic adaptations can arise. The consequence of these mutations influences subsequent risk of progression to MDS/AML, and the stochastic nature of these acquired changes on the cellular level may explain some of the variability in penetrance of MDS/AML in families with inherited bone marrow failure and MDS predisposition syndromes.
Distinct somatic genetic rescue mechanisms can be present within the same family and impact clinical course. Representative pedigrees of SAMD9/9L syndrome (A) and short telomere syndromes (B) showing that within 1 family carrying the same germline mutation, different somatic adaptations can arise. The consequence of these mutations influences subsequent risk of progression to MDS/AML, and the stochastic nature of these acquired changes on the cellular level may explain some of the variability in penetrance of MDS/AML in families with inherited bone marrow failure and MDS predisposition syndromes.
Somatic TERT promoter and POT1 mutations protect against MDS/AML in the short telomere syndromes
Some of the earliest identified rescue mechanisms were in the short telomere syndromes. As with the adaptive genetic mechanisms seen in SAMD9/9L syndromes, the initial reports identified mechanisms that directly repaired the germline mutant allele itself via mitotic recombination with the wild-type allele, somatic second-site mutations, or back mutations.57-59 In adults with heterozygous germline deletions in the telomerase RNA (TR) gene (located on chromosome 3q), mitotic recombination with the wild-type allele, which lead to uniparental isodisomy of 3q, resulted in reversion to the wild-type genotype in the affected blood cells.58 This was documented in 4 of 12 individuals with small (1-4 nucleotide) deletions in TR but has not been replicated in other cohorts.58 In an adult with a germline gain-of-function mutation in TINF2, a somatic second-site frameshift deletion in cis provided an advantage and abolished the BMF phenotype.57 In a child with dyskeratosis congenita due to germline mutation in the 5′ UTR of DKC1, acquired back mutation led to correction of the germline mutation and normalization of dyskerin in the cells bearing the reversion.59
More recently, deep sequencing has uncovered multiple mechanisms of SGR that may be seen in up to 30% of older adults with short telomere syndromes.49 The most common are somatic mutations in canonical sites in the promoter region of TERT.49,60,61 These TERT promoter mutations are a common telomere maintenance mechanism in many malignancies and create a de novo ETS transcription factor binding site, which upregulates transcription of the TERT allele.62 In patients with germline loss-of-function TERT mutations, these somatic TERT promoter mutations occur on the wild-type allele (i.e; in trans with the germline mutation) and thus preferentially upregulate expression of wild-type TERT (Figure 4).60,61
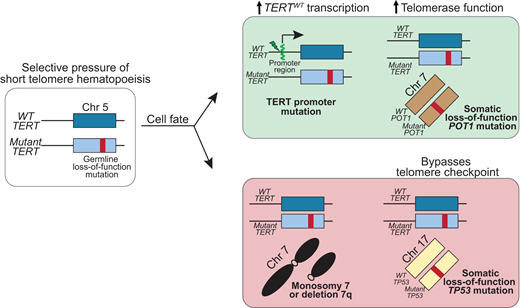
Somatic genetic rescue mutations that promote telomere elongation are protective of developing MDS/AML in the short telomere syndromes.TERT promoter mutations arise in trans with the germline TERT loss-of-function mutation and upregulate transcription of the wild-type TERT. POT1 mutations are a more universal rescue mechanism with no preference for germline mutant gene, and POT1 loss-of-function mutation improves telomerase access to the telomere and/or increases telomerase processivity. Monosomy 7 and biallelic TP53 mutations, also shared risk factors in other BMF/MDS syndromes, are associated with progression to MDS/AML. Chr, chromosome; WT, wild type.
Somatic genetic rescue mutations that promote telomere elongation are protective of developing MDS/AML in the short telomere syndromes.TERT promoter mutations arise in trans with the germline TERT loss-of-function mutation and upregulate transcription of the wild-type TERT. POT1 mutations are a more universal rescue mechanism with no preference for germline mutant gene, and POT1 loss-of-function mutation improves telomerase access to the telomere and/or increases telomerase processivity. Monosomy 7 and biallelic TP53 mutations, also shared risk factors in other BMF/MDS syndromes, are associated with progression to MDS/AML. Chr, chromosome; WT, wild type.
A diverse number of additional SGR mechanisms appear to converge on enhancing telomerase levels and function.49 For example, loss-of-function mutations in POT1 appear to offset germline defects in TERT as well as multiple other genes. In patients with inherited mutations in TR or related pathways, somatic mutations that minimize telomerase RNA degradation or restore telomerase RNA levels appear to be restricted to that group of patients. Some of these mutations are also identical to those that are seen in cancer, but in the short telomere syndromes, they appear to avert the telomere crisis and to be protective against MDS/AML. The evidence that these somatic rescue mutations are beneficial when at high allele frequency (VAF > 10%) is that they were not seen in patients with short telomere MDS/AML and are generally mutually exclusive with cytogenetic abnormalities, such as monosomy 7.9,49 Multiple SGR mechanisms can coexist in separate clonal populations in an individual (Figure 3B).49 TERT promoter and POT1 mutations are commonly included on clinical next-generation sequencing (NGS) panels and may provide clinically relevant information in assessing the course of telomere-mediated BMF in older adults.
Somatic genetic rescue in Shwachman-Diamond syndrome
Shwachman-Diamond syndrome is an autosomal recessive disorder that, in 90% of cases, is caused by homozygous or compound heterozygous mutations in the SBDS gene, located on chromosome 7q11.63 Biallelic loss-of-function mutations in SBDS lead to low levels of SDS protein, which is required along with EFL1 to remove eIF6 from the 60S ribosome subunit prior to ribosome maturation; hence, these mutations result in defective ribosome assembly.64,65 Interstitial deletion of chromosome 20q (del20q), which encompasses the EIF6 gene, and isochromosome i(7)(q10) (i7q) were observed over a decade ago in the blood and bone marrow in approximately 20% of patients with biallelic SBDS mutations.66,67 Studies of cases with the common, hypomorphic SBDS germline mutation, c.258 + 2 T>C, in a compound heterozygous state, showed that i7q nonrandomly occurs on the same allele containing c.258 + 2 T>C. The c.258 + 2 T > C mutation results in expression of an alternatively-splice truncated protein in addition to a scant amount of normal protein.68,69 Thus, when this particular mutant gene is duplicated, relatively more protein is produced. Large del20q clones (>10% of bone marrow cells) had been associated with a benign course and low risk of progression to MDS.70,71 However, a recent registry-based cohort study found del20q and i7q were transient findings in up to one-quarter of patients.72 Further, there are reports of MDS/AML in some individuals with these cytogenetic alterations, although the del20q and i7q were often absent from the malignant clone. Thus, the clinical significance of these as low risk or protective findings remains less clear.
Using ultradeep sequencing of two independent cohorts, somatic mutations in EIF6 itself are found in up to 60% of individuals with Shwachman-Diamond syndrome due to biallelic SBDS mutations.47,73 Expression of the EIF6 mutations in leukemia cell lines resulted in either decrease in eIF6 protein amount or disruption in eIF6 and 60S binding. When expressed in SBDS- deficient human CD34+ cells, inactivating EIF6 mutations resulted in increased colony formation and improvement in the ribosome assembly defect supporting a functional compensation for SBDS deficiency.47 The presence of somatic EIF6 mutations was also not associated with severe BMF, leukemic transformation or, by single cell sequencing, TP53 co-mutation.47 Additional longitudinal follow-up is needed to further assess the impact of EIF6 mutations on MDS/AML risk, particularly since most mutations were present at low abundance (<1% VAF, below the limit of clinical detection).
Diagnostic evaluation for SGR events in clinical practice
The identification of these mechanisms has revealed the importance of incorporation of somatic genomic testing into routine hematologic surveillance for individuals with BMF/MDS syndromes. Many SGR mechanisms described here can be assessed on commonly-utilized clinical molecular tests, while others require dedicated testing (Table 3). Hematologic malignancy somatic NGS panels can detect second-site reversion mutations, indirect SGR mutations, and second-hit mutations, as long as the genes or regions of interest are captured by the particular panel. Genomic microarray is needed to detect copy number alterations, such as CN-LOH indicative of uniparental isodisomy, which is particularly important in SAMD9/9L syndromes among others (Table 2).
Clinically available molecular tests to detect common adaptive mutations in patients with inherited bone marrow failure and MDS predisposition syndromes
| Somatic genetic rescue mechanism | Molecular test |
|---|---|
| Somatic 2nd site mutations Other direct reversion mutations | Somatic NGS of gene of interest* |
| Indirect rescue mutations such as: TERT promoter** POT1 | Somatic NGS of gene of interest* |
| Clonal cytogenetic abnormalities such as: Interstitial deletion 20q Isochromosome 7q Monosomy 7 | Conventional karyotype FISH |
| Copy neutral-LOH to detect UPD | Chromosomal microarray |
| Somatic genetic rescue mechanism | Molecular test |
|---|---|
| Somatic 2nd site mutations Other direct reversion mutations | Somatic NGS of gene of interest* |
| Indirect rescue mutations such as: TERT promoter** POT1 | Somatic NGS of gene of interest* |
| Clonal cytogenetic abnormalities such as: Interstitial deletion 20q Isochromosome 7q Monosomy 7 | Conventional karyotype FISH |
| Copy neutral-LOH to detect UPD | Chromosomal microarray |
Whether the gene of interest is included in clinically available somatic NGS panels is gene dependent. Clinical assays to date also have a higher threshold for detection of somatic mutations than the more sensitive deep sequencing approaches utilized in the research setting.
Somatic genetic rescue mutations at three canonical sites in the TERT promoter are found in patients with short telomere syndromes: c.-124, c.-146 and c.-57. That latter site, c.-57 C>T, is less commonly included in clinical somatic mutation panels.
FISH, fluorescence in situ hybridization.
Concordance of mutation detection between peripheral blood and bone marrow, particularly for larger clonal changes (VAF >5%), may allow screening and monitoring of SGR mechanisms from peripheral blood in some settings.74 For example, clinically relevant TERT promoter and POT1 mutations can be reliably detected from the peripheral blood in individuals with short telomere syndromes.49 Our practice is to use peripheral blood NGS to screen older adults with short telomere syndromes (at least over age 40) for protective SGR mutations. Presence of a TERT promoter mutation with VAF >10% identifies an individual at lower risk of progression to MDS/AML for whom monitoring via serial blood counts and peripheral blood NGS is appropriate. Additional studies and longer follow-up are needed to more definitively test the predictive utility of SGR testing in clinical settings and to refine actionable thresholds for each specific BMF/MDS syndrome.
Summary and future directions
The availability of higher resolution sequencing has uncovered a pattern of rescue hematopoiesis in individuals with inherited BMF/MDS syndromes. The adaptative rescue mechanisms identified to date are exquisitely disease-specific and driven by the underlying biology; their detection in seemingly sporadic cases should prompt consideration of an underlying germline BMF/MDS predisposition, if not already identified. The recent evidence demonstrates that adaptive SGR mechanisms are often more common than the overall prevalence of MDS/AML. These emerging observations identify a leukemogenesis paradigm that is shared across multiple BMF/MDS syndromes whereby, in the highly replicative environment of hematopoiesis, disease-specific adaptive modifications arise that overcome the inherited defect. More rarely, maladaptive responses emerge with increased leukemogenic potential. Future studies aimed at enhanced detection of leukemogenic alterations have the potential to improve clinical outcomes by enabling preemptive intervention in the highest risk patients.
Acknowledgments
The author is thankful to Dr. Mary Armanios for helpful discussions and critical reading of the manuscript and figures and to Jennifer Fairman for help with an illustration. The author is appreciative of her patients and their families for their invaluable contributions to understanding these disorders. Dr. Schratz is supported by a grant from the National Institutes of Health (NIH), National Heart, Lung, and Blood Institute K08HL163468 and the Commonwealth Foundation to the Sidney Kimmel Comprehensive Cancer Center (SKCCC). SKCCC is supported by NIH P30CA006973.
Conflict-of-interest disclosure
Kristen Schratz: no competing financial interests to declare.
Off-label drug use
Kristen Schratz: Nothing to disclose.
