

Abstract
In case 1, a 14-month-old male child with sickle cell disease (SCD) was referred for evaluation for an allogeneic hematopoietic stem cell transplant (HCT). The patient had a history of dactylitis 3 times in his first year of life and febrile episodes twice at the consult. His 4-year-old sister was found to be human leukocyte antigen (HLA) identical. The patient was started on hydroxyurea (HU) at 2.5 years of age. His parents again sought consultation when he was 5 years old because of concerns about his medical condition. At the time, the patient had experienced 2 vaso-occlusive pain episodes (VOEs) requiring hospitalization during the previous 2 years. He had also experienced intermittent pain crises requiring rest at home for 2 to 3 days. The child has not attended school in person due to the COVID-19 pandemic. The family is considering HCT but is ambivalent about it because of potential toxicity. In case 2, an 8-year-old female child is 3 years out from HCT for SCD from her HLA-identical sibling. Before HCT, despite receiving HU, she had experienced >5 VOEs requiring hospitalization and 2 episodes of acute chest syndromes in the previous 3 years. She had also been missing almost 50 days of school days each year. After HCT, she is now attending school regularly and participating in all normal age-appropriate activities. The parents believe that HCT has been transformative in their child's life.
Learning Objectives
Understand therapeutic options for a young child with SCD
Describe the current outcomes of MSD HCT
Critically appraise the role of MSD HCT for SCD in young children
Introduction
Sickle cell disease (SCD) is an autosomal recessive inherited hemoglobinopathy. Over 300 000 affected babies are born each year with SCD, which is expected to increase to over 400 000 a year by 2050.1,2 Although the overwhelming majority of patients live in Africa, India, or the Middle East, growing numbers of individuals with SCD are encountered in areas that were historically not endemic for malaria because of population migrations. In the United States, it is estimated that 100 000 individuals are affected with SCD.2-4 SCD is associated with a significant risk of morbidity and premature mortality.5 Thus, SCD is a significant public health problem worldwide that disproportionately affects populations residing in resource-poor settings. Hematopoietic cell transplantation (HCT) can ameliorate long-term SCD-related morbidity and its consequences.
Outcomes with comprehensive care and disease-modifying therapy for SCD
Advances such as newborn screening, penicillin prophylaxis, pneumococcal immunization, and comprehensive care have improved survival in children with SCD. Hydroxyurea (HU) has been proven to effectively decrease the frequency of vaso-occlusive pain episodes (VOEs), acute chest syndrome, frequency of transfusions, and death in multiple randomized controlled trials.6 In addition, HU is an effective substitute for chronic transfusions to prevent primary stroke among high-risk pediatric patients with SCD who have abnormal transcranial Doppler (TCD) flow velocity.5 Observational studies have demonstrated a relationship between HU use and decreased rates of hospitalization and blood transfusions.6 Thus, this relatively inexpensive orally administered medication can be used globally to ameliorate SCD complications and improve survival. Nouraie et al7 reported the results of the pulmonary hypertension and the hypoxic response in sickle cell disease study that estimated that the survival of a cohort of children, adolescents, and young adults with SCD was 99% to 18 years, and this declined to 94% at 25 years. Stroke was the most common cause of death. Elevated systolic pulmonary artery pressure as reflected in triscupid regurgitant jet velocity (TRV) ≥2.7 m/s, high serum ferritin concentration, low forced expiratory velocity (FEV1)/forced vital capacity, and high neutrophil count were biomarkers of increased risk of death, predominantly after the transition to adulthood. Recently, newer disease-modifying agents approved by the US Food and Drug Administration (FDA) offer added potential to improve outcomes for patients with SCD. The new disease-modifying drugs include voxelotor, which binds to the N-terminus of sickle hemoglobin to stabilize oxygenated sickling and prevent sickling8 ; L-glutamine, which reduces oxidative stress and could result in fewer episodes of VOE9 ; and crizanlizumab, which acts on cytoadherence of sickle red blood cells, leukocytes, and platelets to the endothelium.10 These drugs offer the potential for further ameliorating complications of SCD and provide an alternative for HCT for disease modification. However, in the absence of long-term data, we do not know how the additional FDA-approved SCD therapies will modify the clinical history of patients with SCD.
The improvement in survival of children with SCD between 1979 and 2005 seemed to mirror a similar decrease in adult survival during the same period.11 Although individual centers in high-resource settings report a median survival of 58 to 67 years overall, there has been little improvement in survival for adults with SCD.12 In adults, there is a progressive loss of pulmonary FEV1 at 49 cc/y.13 More than 61% of patients with SCD aged 18 to 30 years develop either micro- or macroalbuminuria.14 There is a worsening change in tricuspid regurgitant jet velocity over time.15 There is neurocognitive decline and worse performance IQ, processing speed, cognitive function, and executive function with age.16 Acute-on-chronic morbidity leads to reduced health-related quality of life (HRQOL), depression and anxiety, diminished educational and occupational fulfillment, and premature mortality, contributing to health care costs..2,17,18 Disease-modifying therapies and supportive care can improve outcomes but place a substantial burden of care, requirement of medication adherence, and interruption of daily life.17
Thus, a child receiving comprehensive care in high-resource settings has an estimated 99% survival into adulthood. However, there may be progressive organ damage, impaired quality of life, considerable morbidity in childhood, and risk of premature mortality in adulthood. However, it is crucial to recognize that it is not yet known if the additional FDA-approved SCD therapies will modify the clinical history of patients with SCD in adulthood.
Outcomes after matched sibling donor bone marrow transplant
HCT for SCD was first performed in 1984 from a human leukocyte antigen (HLA) matching sibling donor (MSD) to treat acute myeloid leukemia in a patient who also had SCD and paved the way for a curative option for SCD. The early transplant series using myeloablative conditioning, such as a transplant for malignant diseases, showed overall survival (OS) and event-free survival (EFS) in the range of 77% to 95% and 93% to 95% following HCT from an HLA MSD, respectively.18,19 In the past decade, there has been further improvement in the outcomes of HCT for SCD, with OS and EFS in the range of 94% to 100% and 91% to 100%, respectively, with a minimum risk of mortality with reduced-intensity/toxicity regimens.20-24 An international, retrospective, registry-based analysis reported an OS of 92.9% and EFS of 91.4% in 1000 children who underwent HCT between 1986 and 2013.21 On multivariate analysis, survival was better with bone marrow than peripheral blood stem cells graft and those who underwent transplantation after 2006.21 Cappelli et al,22 in a study of recipient age as a predictor of the outcome of MSD HCT, reported an OS and EFS of 100% and 93%, respectively, in children 0 to 5 years of age. Bernaudin et al25 reported outcomes of MSD HCT for SCD from 1988 to 2012 and described the improvement in outcome since 2005. They noted that for MSD since 2005, OS is 98.6% and EFS is 97.4%. The use of reduced-intensity or nonmyeloablative conditioning strategies such as combining alemtuzumab with fludarabine and melphalan,26 as well as low-dose total body irradiation (300 cGy),20,27 has been associated with high OS and EFS in both adults and children.
The risk-benefit trade-off in HCT for SCD
The decision to proceed with curative options for SCD represents a complex risk-benefit trade-off. On one hand, survival to adulthood is almost universal in children receiving the best comprehensive care. The disease, however, progresses in the majority to cumulative organ damage, morbidity, and mortality in adulthood. On the other hand, following HLA MSD HCT in young children, there is a high probability of survival, with a potential for alleviating symptoms related to SCD, stabilizing organ function, and the possibility of the patient being able to live relatively everyday life. But HCT itself is associated with substantial complications in the short term, a significant burden of care that can be disruptive to the patient and caregivers, and a small risk of mortality.
Furthermore, the long-term sequelae of HCT, such as graft-versus-host disease, infertility, and subsequent malignancy, could be unintended consequences of this therapy (Figure 1). This decisional dilemma is also further compounded by the patient's age, the severity of the current clinical course, the type of donor available, and the availability of other treatment options. Thus, parents considering HCT for their young child face multiple complex trade-offs: the primary trade-off for a family of a 5-year-old is survival of the child in the decade when they age from 5 to 15 years. Although no study has compared HCT with standard of care in children with SCD, published studies suggest that survival to the next decade for a 5-year-old child with SCD following standard of care or MSD HCT may be comparable. However, a parent may also consider other trade-offs such as the outcomes of HCT in childhood vs the potential for morbidity and mortality in adulthood, HRQOL with or without HCT, and curative HCT vs chronic GVHD, infertility, cardiovascular risk, and subsequent malignancy.28 How families resolve these decisional trade-offs is likely to be highly individualized and dependent on several factors unique to the circumstances of the patient and family. To assist the hematologist in the conversation with the family of a young child with SCD who has an HLA-identical sibling, we summarize the available evidence as answers to critical questions that may arise in the minds of patients and their caregivers as they contemplate HCT.
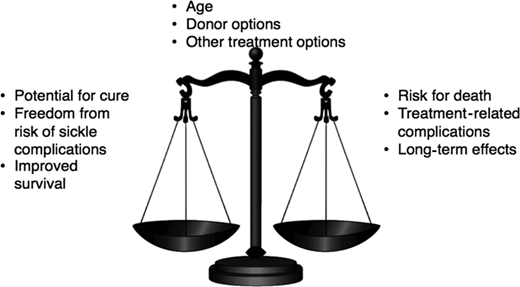
Who has been offered HCT for SCD?
HCT for SCD has been offered to patients with severe SCD- related complications. Indications for HCT include stroke, the need for chronic blood transfusions to prevent SCD-related complications, and severe disease complications such as recurrent VOE, recurrent acute chest syndrome, sickle nephropathy, or retinopathy that at least affects vision in 1 eye or osteonecrosis.18,29 In early clinical trials of HCT for SCD, the most common indications were stroke in 57% of patients and frequent VOEs in 23% of patients. The most common indication for HCT recently has been recurrent VOE in more than 70% of the cases.21 The change in the most frequent indication for HCT may reflect the decrease in the incidence of stroke because of better comprehensive care, screening for stroke risk, and primary prevention of stroke by the institution of chronic blood transfusion. It is also possible that physicians and patients are more accepting of HCT for individuals with recurrent VOE. Utilization of health care for recurrent VOE is being used as a marker of disease severity and a predictor of long-term survival in adults. Yet, health care utilization for VOE is not an accurate measure of pain burden because many VOE episodes are managed at home. Thus, some patients may have chronic disabling pain but may not access health care for various reasons.30 There is, therefore, a need to better estimate pain burden to inform risk-benefit stratification in consideration of HCT for SCD. An ongoing clinical trial is studying HCT from HLA matched sibling HCT for children with the less severe clinical phenotype (ClinicalTrials.gov NCT04018937).
Is it possible to offer MSD HCT to every child with SCD?
The applicability of HCT is limited because only 14% to 18% of patients with SCD have an available HLA-identical family donor.31 Most sibling donors can have sickle cell trait. However, HCT from a donor with sickle cell trait is safe and effective.31,32 Clinical trials are evaluating alternate donor HCT. An estimated 19% of the patients may find a suitable HLA matched unrelated donor,32 and most are likely to find a haploidentical family donor. If the safety and efficacy of alternate donor HCT are proven, a more significant proportion of patients may be provided HCT. Thus, only a small minority of patients can be offered HCT from an HLA- identical family donor.
What is the best time to perform HCT?
Age at HCT is the most critical determinant of outcome in patients with SCD with an available HLA MSD. Patients receiving HCT who are younger than 5 years have an EFS of 93% and an OS of 100% compared with 89% and 95% for those aged 5 to 10 years and 81% and 88% for those older than 15 years, respectively.22 Gluckman et al21 report that for every 1-year increment in age, there was a 9% increase in the hazard ratio for treatment failure—that is, graft failure or death. Similarly, for every 1-year increment in age, there was a 10% increase in the hazard ratio for death.22 Risk of graft failure, grade II to IV acute graft-versus-host disease (GVHD), and chronic GVHD are lowest in children younger than 5 years at HCT. Gluckman et al21 reported from the European Society for Blood and Marrow Transplantation registry that many centers have diverse experience in performing HCT for SCD. This is mirrored by the report by Bernaudin et al23 in a single-center study with a 5-year EFS of 97.9% (95% confidence interval, 95.5%-100%). Brazauskas et al24 developed a risk scoring system that included age at HCT and donor type to predict EFS after HCT for SCD. They concluded that patients 12 years and younger with an HLA-identical family donor have the lowest risk.
Another primary consideration in the timing of HCT for SCD is the presence or imminence of organ damage. Therefore, HCT should preferably be offered to patients who have been predicted to have a poor outcome but have not experienced severe organ damage to avoid serious side effects associated with HCT procedures. However, SCD organ damage is insidious, with progressive loss of FEV1 at 49 cc/y,13 61% of patients with SCD aged 18 to 30 years developing either micro- or macroalbuminuria,14 worsening changes in tricuspid regurgitant jet velocity over time,15 and neurocognitive decline with poor performance IQ, processing speed, cognitive function, and executive function.16 Because overall outcomes of MSD HCT for SCD are best when HCT is performed at age 5 years or younger, the consensus opinion of an European Society for Blood and Marrow Transplantation expert panel is to consider MSD at a young age.33
What about long-term side effects and quality of life after HCT compared with patients with SCD who have not received curative therapies?
Improvement in HRQOL outcomes in patients with SCD following HCT has been reported in multiple studies.34-39 This includes significant improvement in physical, emotional, psychosocial, general health, bodily pain, pain interference, and vitality domains of HRQOL. It has also been reported that cardiac, neurologic, and neuropsychologic functions stabilize after HCT.19 In the most recent report, for children with SCD requiring chronic transfusion because of persistently elevated TCD velocities, MSD HSCT was significantly associated with lower TCD velocities at 1 year compared with standard care.25 One of the significant risks following HCT is gonadal damage and infertility.19 Fertility preservation by sperm banking or cryopreservation of oocytes can potentially mitigate the risk of infertility. Ovarian tissue cryopreservation in prepubertal females is now considered the standard of clinical care. Experimental procedures such as testicular tissue cryopreservation are still under investigation.40,41 On the other hand, HU also adversely affects male and female fertility in patients with SCD.42,43 The risk of subsequent malignancy following HCT remains small.41,44,45 Late effects of HCT include cardiovascular, pulmonary, and endocrine complications; dysfunction of the thyroid gland, gonads, liver, and kidneys; infertility; iron overload; bone diseases; infection; malignancy; and neuropsychological effects.46 Most of these late effects have been studied in patients undergoing HCT for cancers or bone marrow failure syndromes. There is a need to generate data on the long-term outcome of HCT for SCD to address this element of uncertainty in the decision-making regarding HCT for SCD.47,48
Is HCT cost-effective?
Although the cost-effectiveness of HCT for SCD is still being evaluated, early data suggest HCT is associated with decreased hospitalization, health care utilization, and opioid utilization.49 In adults with SCD, Saraf et al50 reported lower health care costs by the second year post-HCT (median of $16 281 vs $64 634 pre-HSCT [P = .01] vs $54 082 in the standard-of-care group [P = .05]). They reported a median reduction of –$20 833/patient/y (interquartile range, –$67 078 to +$4442/patient/y) in health care costs compared to pre-HCT in the second year post-HSCT (P = .05). In children, Arnold et al,51 while reviewing 2 years pre- and post-HCT data, reported significant reductions in admissions (P < .001), length of stay (P < .001), and cost (P = .008) for HCT patients who received their HCT before the age of 10 years.
Should we wait for gene therapy clinical trials or for gene therapy to be approved instead of considering allogeneic HCT?
Early phase clinical trials of autologous transplantation of hematopoietic progenitors modified by gene addition or gene editing hold the promise of long-term disease amelioration without the need for an allogeneic donor and associated risk of GVHD. Gene therapy studies have so far enrolled patients 12 years or older. Future studies may offer enrollment to younger children. The studies typically exclude children who have an available HLA MSD. The current clinical hold of the Hgb-206 gene therapy trial (ClinicalTrials.gov: NCT02140554) after 2 participants developed acute myeloid leukemia underscores the fact that there remain many unknowns in this experimental therapy.52-58 Thus, until a viable, safe strategy has been implemented for the gene therapy approach, HCT is still considered the best curative option for young children with SCD if they have an available HLA MSD, particularly with reduced-intensity/toxicity conditioning regimens. There have been no comparative trials of allogeneic HCT and autologous gene therapy for any other diseases. Thus, further studies are required to determine the relative safety and efficacy of the 2 treatment modalities.
Are there individual differences in how patients and families consider the information and make decisions about HCT?
The decision for HCT for SCD involves complex decisional trade-offs. Parents deciding to proceed with HCT face many dilemmas such as the unpredictable onset and progression of SCD complications, repeated need to seek health care with SCD, possible poor quality of life with SCD, the imminence of making a major therapeutic decision, and concerns about the long-term consequences of the disease. The feeling that the disease burden has become unacceptable is frequently cited as a trigger for the parental decision.28,59,60 The availability of an HLA MSD may be viewed as providential by some parents. Parents vary greatly in their willingness to accept risks associated with HCT to achieve potential benefits. HCT-associated morbidity, including mucositis, GVHD, risk of death, and infertility, is a significant reason that gives families pause in accepting HCT options. Overall, families face substantial decisional conflict.60 Some patients or caregivers would not consider HCT at any level of risk.61,62 Almost three-fourths of patients report being willing to take a modest risk of ≥5% mortality risk, whereas 57% are ready to take ≥10% risk of GVHD. Some parents and caregivers may also have difficulty making decisions about HCT for and on behalf of a child who is currently doing well and may have to deal with long-term sequelae of HCT.28 However, following successful HCT, parents do not report decisional regret even if their child has had severe HCT-related complications.59 Thus, the decision to undergo HCT is highly individualized and is driven by the perception of disease severity; the values and preferences of patients, caregivers, and their physicians regarding disease-modifying options; and acute and long-term sequelae of HCT.28,63
Summary and recommendations
Young children with SCD have an excellent outlook for survival to adulthood, and new drugs may further improve outcomes. However, many children may have severe disease manifestations, organ damage, and impaired quality of life. Disease progression may be unpredictable in adulthood, with severe consequences for the patient. HCT from HLA MSDs in young children is associated with excellent OS and EFS, stabilization of organ function, decreased health care utilization, and improved quality of life. These benefits come at the cost of acute morbidity, the substantial short-term burden of care, a small risk of HCT-related mortality, late sequelae such as infertility, and a slight increase in the risk of subsequent malignancy.
Furthermore, with increasing age, there is a decline in the outcomes of HCT. Parents and caregivers approach these complex trade-offs in ways unique to their life situations and experiences and their perception of disease burden. We, therefore, recommend routine HLA typing of full siblings and consultation with a physician knowledgeable about HCT as 1 component of comprehensive and supportive care, which includes the use of current disease-modifying therapies. The complexity of decision-making regarding HCT necessitates a balanced discussion of the decision with the family in shared decision-making about HCT. Communication about the patient between the hematology and HCT teams is also likely to be beneficial. Families may also benefit from a consultation with support groups and peers who may have navigated a similar decisional dilemma.
The 8-year-old patient described in the second vignette illustrates such a process. The patient had severe disease manifestations and impaired quality of life. Routine HLA typing had revealed the availability of an HLA MSD. The family engaged in the consultative and decisional process and decided to proceed with HCT. Following HCT from the HLA MSD, the child is currently leading a normal life.
The toddler in the first vignette is beginning to manifest complications of SCD. The parents, however, are ambivalent about HCT. We would recommend continuing comprehensive medical care, including the addition of disease-modifying therapy and support for treatment adherence and psychosocial functioning. If the family were to elect to proceed with HCT in the future, we recommend that HCT be offered in the context of a multicenter clinical trial. Active consideration must also be given to providing fertility preservation procedures pre-HCT and long-term follow-up post-HCT to monitor for late effects of treatment.
Conflict-of-interest disclosure
Niketa Shah: no competing financial interests to declare.
Lakshmanan Krishnamurti: no competing financial interests to declare.
Off-label drug use
Niketa Shah: no off-label drug use has been discussed.
Lakshmanan Krishnamurti: no off-label drug use has been discussed.
