

Abstract
Inherited microcytic anemias can be broadly classified into 3 subgroups: (1) defects in globin chains (hemoglobinopathies or thalassemias), (2) defects in heme synthesis, and (3) defects in iron availability or iron acquisition by the erythroid precursors. These conditions are characterized by a decreased availability of hemoglobin (Hb) components (globins, iron, and heme) that in turn causes a reduced Hb content in red cell precursors with subsequent delayed erythroid differentiation. Iron metabolism alterations remain central to the diagnosis of microcytic anemia, and, in general, the iron status has to be evaluated in cases of microcytosis. Besides the very common microcytic anemia due to acquired iron deficiency, a range of hereditary abnormalities that result in actual or functional iron deficiency are now being recognized. Atransferrinemia, DMT1 deficiency, ferroportin disease, and iron-refractory iron deficiency anemia are hereditary disorders due to iron metabolism abnormalities, some of which are associated with iron overload. Because causes of microcytosis other than iron deficiency should be considered, it is important to evaluate several other red blood cell and iron parameters in patients with a reduced mean corpuscular volume (MCV), including mean corpuscular hemoglobin, red blood cell distribution width, reticulocyte hemoglobin content, serum iron and serum ferritin levels, total iron-binding capacity, transferrin saturation, hemoglobin electrophoresis, and sometimes reticulocyte count. From the epidemiological perspective, hemoglobinopathies/thalassemias are the most common forms of hereditary microcytic anemia, ranging from inconsequential changes in MCV to severe anemia syndromes.
Learning Objectives
Understand the criteria to define microcytic anemias and their differential diagnosis: acquired and congenital/hereditary
Understand the roles of hepcidin and erythroferrone in iron abnormalities in hereditary microcytic anemias (IRIDA, atransferrinemia, and DMT1 deficiency)
Introduction and classification of microcytic anemias
Erythropoiesis is a complex and sophisticated process through which hematopoietic cells differentiate into erythroid progenitors and then to reticulocytes that become mature red blood cells (RBCs) in the peripheral blood. Approximately 2 × 1011 erythrocytes are released daily and replace the 2 × 1011 erythrocytes removed daily from the peripheral circulation mainly through the reticuloendothelial system, particularly the spleen. The late phase of erythropoiesis is characterized by ≥4 distinct events: (1) progressive reduction of cell volume, (2) condensation of chromatin, (3) synthesis of hemoglobin (Hb) and (4) organization of the red cell membrane.1
Microcytic anemia is the most common form of anemia, both in childhood and in adulthood. Microcytic anemias are highly heterogeneous, and they may be either acquired (mostly due to iron deficiency) or inherited. These latter forms may be present at or around birth; however, in the vast majority of cases, the clinical appearance is delayed, and the diagnosis is achieved during childhood.2
Microcytic anemia is defined as a reduced Hb synthesis associated with RBC mean corpuscular volume (MCV) <80 fL during adulthood.2 Of note, MCV is lower in childhood than in adulthood, and a nutritional iron deficiency may account for the appearance of congenital microcytic anemia (present at or around birth) that can be misdiagnosed as an inherited form of microcytic anemia.3
Inherited microcytic anemias embrace a wide spectrum of conditions associated with different pathogenic mechanisms. Indeed, these conditions can be broadly classified into 3 subgroups: (1) defects in globin chains (hemoglobinopathies and thalassemias), (2) defects in heme synthesis (truly, protoporphyrin IX deficiency), and (3) defects in iron availability or iron acquisition by the erythroid precursors (Figure 1). These conditions are characterized by decreased availability of Hb components (globins, iron, and heme) that in turn causes a reduced Hb content in RBC precursors with subsequent delayed erythroid differentiation. As a main consequence, it has been hypothesized that erythroid precursors undergo an additional mitotic cycle to overcome the reduced Hb concentration, with subsequent production of erythrocytes with reduced cellular volume.4 Moreover, the heme-regulated inhibitor, a key heme-binding protein that senses intracellular heme concentrations to balance globin protein synthesis with the amount of heme available for Hb production, plays a crucial role in iron/heme deficiency and β-thalassemia. Indeed, the repression of globin mRNA translation by heme-regulated inhibitor reduces proteotoxicity and permits the expression of ATF4 protein, which plays a pivotal role in terminal erythropoiesis by maintaining oxidative homeostasis and mitochondrial functions. Furthermore, ATF4 represses mammalian target of rapamycin complex 1 (mTORC1) signaling and provides a feedback mechanism to attenuate erythropoietin-mTORC1–stimulated ineffective erythropoiesis in iron deficiency anemia (IDA). Inhibition of mTORC1 also improves anemia and promotes erythroid differentiation of Foxo3−/−, β-thalassemic, and mice with sickle cell disease.5 On the contrary, in atransferrinemia, DMT1 deficiency, and ferroportin disease, microcytic anemia is associated with iron overload because the iron is not made available for erythropoiesis and accumulates in some organs.6 This review focuses on congenital and hereditary microcytic anemias due to impaired synthesis of globin chains (thalassemias) and heme or due to iron metabolism defects resulting in either iron deficiency or iron overload.7
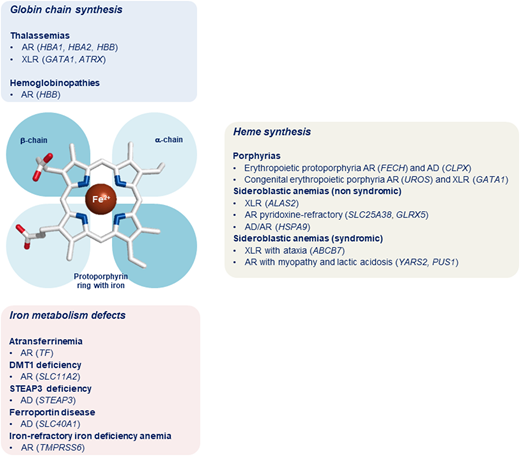
Classification of microcytic anemias. In the middle of the figure is a schematic representation of the prosthetic group of hemoglobin with a protoporphyrin ring (with iron) and the globin chains. In the top panel (light blue) is the classification of microcytic anemias caused by alterations of globin chain synthesis. In the right panel (gray) is the classification of microcytic anemias caused by impairment of heme synthesis. In the bottom panel (pink) is the classification of microcytic anemias caused by iron metabolism defects. AD, autosomal dominant inheritance; AR, autosomal recessive inheritance; XLR, X-linked recessive inheritance.
Classification of microcytic anemias. In the middle of the figure is a schematic representation of the prosthetic group of hemoglobin with a protoporphyrin ring (with iron) and the globin chains. In the top panel (light blue) is the classification of microcytic anemias caused by alterations of globin chain synthesis. In the right panel (gray) is the classification of microcytic anemias caused by impairment of heme synthesis. In the bottom panel (pink) is the classification of microcytic anemias caused by iron metabolism defects. AD, autosomal dominant inheritance; AR, autosomal recessive inheritance; XLR, X-linked recessive inheritance.
Clinical cases
In this section, we describe 2 patients with hereditary microcytic anemia due to different genetic disorders. The first is a typical iron-refractory iron deficiency anemia (IRIDA) case; the second is a more difficult case with an original clinical suspicion of β-thalassemia subsequently diagnosed as pyruvate kinase deficiency (PKD).
Case 1
The patient in case 1 was an 8-year-old girl with consanguineous parents from Saudi Arabia. Her personal history was characterized by IDA unresponsive to oral iron and partially responsive to parenteral iron administration.8 Her family history highlighted the presence of 2 siblings with a similar clinical picture. The recessive transmission was suggested by parents with normal hematological phenotype, the presence of affected sibling pairs, and consanguinity.
At the time of the diagnosis when the girl was 5 years old, she presented with severe microcytic hypochromic anemia with low serum iron and low transferrin saturation (Tsat). Her complete blood count results included Hb of 8.83 g/dL, MCV of 53.3 fL, mean corpuscular hemoglobin (MCH) of 15.9 pg, mean corpuscular hemoglobin concentration of 29.8 g/dL, red blood cell distribution width (RDW) of 19.6%, and platelet count of 526 × 103/μL. Her iron status was serum iron level of 20 μg/dL, ferritin concentration of 101 ng/mL, Tsat of 3.3%, and soluble transferrin receptor (sTfR) concentration of 5.6 mg/L. Her serum hepcidin levels were above the normal range (5.63 nM; normal range, 3-7 nM).
The finding of the girl’s Hb electrophoresis was normal, and the result of the first-line genetic test for both the common α- and β-thalassemia mutations was negative. Given the girl’s personal and familial history as well as the clinical and biochemical data of the proband, second-line genetic testing was performed with mutational screening of the TMPRSS6 locus. The analysis showed the presence of a homozygous nonsense variant c.1796C>A, p.Ser561*. The analysis of the inheritance pattern confirmed the biallelic inheritance of the variant as well as the presence of the same genotype in the 2 siblings of the proband, both showing the same clinical picture.8
Case 2
The patient in case 2 was a 41-year-old man with nonconsanguineous parents from Naples, Italy. His personal history was characterized by anemia with splenomegaly (spleen measuring 18 cm) since childhood. He had been transfusion dependent from 2 years to 7 years of age. Moreover, he presented with numerous hemolytic crises following infectious episodes. His bone marrow biopsy showed erythroid hyperplasia and a moderate degree of dyserythropoiesis with the presence of binucleated erythroblasts. Target cells and several erythroblasts were present on his peripheral blood smear.
He had first been clinically diagnosed with hereditary spherocytosis, and he had been referred for splenectomy and cholecystectomy at 9 years of age. He was then referred to the Medical Genetics Unit at the Federico II University in Naples for infertility treatment. Indeed, he was diagnosed with hypogonadotropic hypogonadism. At the time of the consultation, he presented with chronic microcytic anemia with signs of hemolysis. His complete blood count results showed an RBC count of 3.20 × 106/μL, Hb concentration of 8.55 g/dL, hematocrit of 27.2%, MCV of 61.5 fL, MCH of 20.5 pg, mean corpuscular hemoglobin concentration of 31.2 g/dL, RDW of 19.4%, platelet count of 279 × 103/μL, and absolute reticulocyte count of 304 × 103/μL (9.5%). The hemolysis signs were total bilirubin of 7.6 mg/dL, unconjugated bilirubin of 6.2 mg/dL, lactate dehydrogenase of 1180 U/L, and undetectable haptoglobin. The patient’s iron balance highlighted a markedly increased level of ferritin (1500 ng/dL).
Globin chain electrophoresis showed increased fetal Hb levels (5%) and increased HbA2 (6%). The patient’s family history highlighted the presence of a first-degree cousin with β-thalassemia major.
Given the patient’s personal and familial histories as well as the clinical and biochemical data of the proband, first-line genetic testing was performed with mutational analysis of the HBB locus. The analysis showed the presence of the common β0-39 mutation in heterozygous state, but no additional variants in the HBB gene were identified. Moreover, deletions/duplications of the HBA locus were excluded, too. Thus, second-line genetic testing was performed using a 71-gene custom panel for hereditary anemias.9 The genomic analysis of the proband highlighted the presence of a well-known pathogenic variant, c.1456C>T, p.Arg486Trp, in the PKLR gene as homozygous. This genotype was compatible with the definitive diagnosis of PKD.
Summary
These 2 paradigmatic cases underline how genetic diagnosis is valuable not only for achieving a correct and conclusive diagnosis but also for guiding possible treatment of patients with anemia. This is mainly true for the treatment of patients with PKD, who require splenectomy for severe forms. Moreover, an allosteric activator of pyruvate kinase enzyme is now available that increases the enzymatic activity in patients with reduced PK enzyme activity.10
Challenges in the diagnosis of hereditary microcytic anemias
Iron metabolism alterations remain central in the diagnosis of microcytic anemia, and, in general, iron status must be evaluated in all cases of microcytosis. Recent years have seen enormous developments in the understanding of iron metabolism, and besides the very common microcytic anemia due to acquired iron deficiency, hereditary iron metabolism abnormalities should be considered in patients with unexplained microcytic anemias (Figure 1). Atransferrinemia (TF gene), DMT1 deficiency (SLC11A2 gene), ferroportin disease (SLC40A1 gene), and IRIDA (TMPRSS6 gene) are now well-established hereditary disorders due to iron metabolism abnormalities, some of which are associated with iron overload.6 Despite this progress, the diagnostic approach for iron metabolism abnormalities remains based on 3 historical tests: serum iron, transferrin (or total iron-binding capacity), and ferritin. Tsat (ie, Tsat is the ratio of serum iron/total iron-binding capacity), serum ferritin, and noninvasive magnetic resonance imaging measurements of liver and heart iron content are other parameters useful in the diagnosis of iron overload conditions.11 The serum sTfR is another marker related to the expansion of erythropoiesis or iron deficiency. The level of hepcidin could be useful in the diagnosis of IRIDA and to decide the therapeutic option for iron supplementation (oral vs IV).12 Tsat/log hepcidin ratio is a parameter that further contributes to the diagnosis of IRIDA.13 A new parameter that is going to enter the diagnostic field is the human serum erythroferrone concentration, which is now available only for research purposes.14,15
Other causes of microcytosis than iron deficiency should be considered. Thus, it is important to evaluate several other RBC and iron parameters in the presence of reduced MCV: MCH, RDW, reticulocyte Hb content, serum iron and serum ferritin levels, total iron-binding capacity, Tsat, Hb electrophoresis, and occasionally reticulocyte blood count. Each of these parameters, as well as the patient’s clinical and family histories, should be taken into account when evaluating a case of microcytic anemia. The latter should be considered to identify transmission modalities and is useful to distinguish between acquired and genetic conditions. Three different steps could be considered for the diagnostic approach to hereditary microcytic anemia (Table 1). In the first evaluation step for a patient with microcytic anemia, the possible presence of a β-thalassemic trait must be excluded. Indeed, microcytosis associated with reduced MCH is the hallmark of β-thalassemia carriers, who usually have normal or very slightly elevated iron parameters and increased HbA2 detected by Hb electrophoresis (high-performance liquid chromatography). RDW is often increased in iron deficiency conditions, such as in DMT1 deficiency, thalassemia, and IRIDA, and it is normal or mildly raised in anemia of chronic disease.16 In patients with IRIDA, the serum iron is low with normal/high serum ferritin, particularly after IV iron therapy has been initiated. Hepcidin is within normal range, but it is inappropriately high in a patient with iron deficiency. The serum iron level is high in the setting of hypotransferrinemia and DMT1 and STEAP3 defects as well as in sideroblastic anemia. Tsat is low in patients with IDA and IRIDA, whereas it is usually elevated in those with sideroblastic anemia. High serum ferritin in the presence of high serum iron and normal transferrin is typical of sideroblastic anemia.
Main features of microcytic anemias
| Acquired conditions | Inherited conditions | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IDA | ACD | BT trait | IRIDA | AHMIO1 | AHMIO2 | EPP1/2 | SIDBA1 | SIDBA2 | SIDBA3 | ASAT | Hypotransferrinemia | |
| Genetic features | ||||||||||||
| Causative gene(s) | — | — | HBB | TMPRSS6 | SLC11A2 | STEAP3 | FECH/CLPX | ALAS2 | SLC25A38 | GLRX5 | ABCB7 | TF |
| Inheritance | — | — | AR | AR | AR | AD | AR/AD | XLR | AR | AR | XLR | AR |
| Hematological and biochemical features | ||||||||||||
| RBC | ↓ | ↓ | ↑ | ↓↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ |
| Hb | ↓ | ↓ | = or ↓ | ↓ or ↓↓ | ↓↓ | ↓↓ | ↓ | ↓ | ↓↓ | ↓↓↓ (age dependent) | ↓ | ↓ |
| MCV | ↓ | ↓ | ↓ | ↓↓ | ↓↓↓ | ↓ | ↓↓ | ↓ | ↓ | ↓↓ | ↓ | ↓↓ |
| RDW | = | = or ↑ | = or ↑ | = | = | ↑ | = | = | ↑ | = | = | = |
| Reticulocytes | ↓ | = or ↑ | = or ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ |
| Tsat | ↓↓ | = | = | ↓↓ or ↓↓↓ | ↑↑ | ↑↑ | ↑ | ↑ | ↑ | ↑ | ↑ | 100% |
| Ferritin | = or ↓ | = | = | = or ↓ | ↑ | ↑ | = | = | = | = | = | = |
| FEP | = or ↑ | = | = | ↑↑ | ↑ | = | ↑↑↑ | = or ↓ | = | = | = or ↓ | = |
| Iron administration | ||||||||||||
| Oral response | Yes | Unpredictable | No | No | No | — | No | No | — | No | No | No |
| Intravenous response | Yes | Unpredictable | No | Yes (not long lasting) | No | — | No | No | — | No | No | No |
| Suggested therapy | Oral iron supplement | Etiologic therapy (EPO, IV iron) | — | — | EPO | — | β-carotene | Vitamin B6 | — | Iron chelation | Vitamin B6 | Plasma; apotransferrin |
| Acquired conditions | Inherited conditions | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IDA | ACD | BT trait | IRIDA | AHMIO1 | AHMIO2 | EPP1/2 | SIDBA1 | SIDBA2 | SIDBA3 | ASAT | Hypotransferrinemia | |
| Genetic features | ||||||||||||
| Causative gene(s) | — | — | HBB | TMPRSS6 | SLC11A2 | STEAP3 | FECH/CLPX | ALAS2 | SLC25A38 | GLRX5 | ABCB7 | TF |
| Inheritance | — | — | AR | AR | AR | AD | AR/AD | XLR | AR | AR | XLR | AR |
| Hematological and biochemical features | ||||||||||||
| RBC | ↓ | ↓ | ↑ | ↓↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ |
| Hb | ↓ | ↓ | = or ↓ | ↓ or ↓↓ | ↓↓ | ↓↓ | ↓ | ↓ | ↓↓ | ↓↓↓ (age dependent) | ↓ | ↓ |
| MCV | ↓ | ↓ | ↓ | ↓↓ | ↓↓↓ | ↓ | ↓↓ | ↓ | ↓ | ↓↓ | ↓ | ↓↓ |
| RDW | = | = or ↑ | = or ↑ | = | = | ↑ | = | = | ↑ | = | = | = |
| Reticulocytes | ↓ | = or ↑ | = or ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ |
| Tsat | ↓↓ | = | = | ↓↓ or ↓↓↓ | ↑↑ | ↑↑ | ↑ | ↑ | ↑ | ↑ | ↑ | 100% |
| Ferritin | = or ↓ | = | = | = or ↓ | ↑ | ↑ | = | = | = | = | = | = |
| FEP | = or ↑ | = | = | ↑↑ | ↑ | = | ↑↑↑ | = or ↓ | = | = | = or ↓ | = |
| Iron administration | ||||||||||||
| Oral response | Yes | Unpredictable | No | No | No | — | No | No | — | No | No | No |
| Intravenous response | Yes | Unpredictable | No | Yes (not long lasting) | No | — | No | No | — | No | No | No |
| Suggested therapy | Oral iron supplement | Etiologic therapy (EPO, IV iron) | — | — | EPO | — | β-carotene | Vitamin B6 | — | Iron chelation | Vitamin B6 | Plasma; apotransferrin |
ACD, anemia of chronic disease; AD, autosomal dominant; AHMIO, anemia, hypochromic microcytic, with iron overload; AR, autosomal recessive; ASAT, sideroblastic anemia with ataxia; BT, β-thalassemia; EPO, erythropoietin; EPP, erythropoietic protoporphyria; FEP, free erythrocyte porphyrin; SIDBA, sideroblastic anemia; XLR, X-linked recessive.
Secondary investigations include erythrocyte zinc protoporphyrin (zinc protoporphyrin or zinc protoporphyrin/heme), plasma hepcidin, and plasma sTfR levels. Bone marrow examination is required to evaluate a potential diagnosis of sideroblastic anemia, although it is not entirely required in patients with a typical presentation and a supportive genetic testing result. Magnetic resonance imaging is generally reserved for monitoring iron in the liver and heart in patients with biochemical evidence of iron overload. High levels of sTfR are found in sideroblastic anemia and in patients with DMT1 and STEAP3 mutations and IRIDA, but not in those with hypotransferrinemia. The serum hepcidin levels are usually reduced in IDA, whereas in patients with IRIDA, hepcidin levels are high or normal.17
A third level of investigations comprises genetic testing, generally reserved for those cases without a satisfactory biochemical explanation. This analysis is used to define atypical forms of anemia and provides useful information for prognosis and treatment. As mentioned in the clinical cases, genetic testing can be performed by either single-gene or multigene analysis using next-generation sequencing approaches. The selection of the appropriate method is based on the evaluation of different criteria: genetic heterogeneity, phenotypic characterization, gene size, and prevalence of the disease. As demonstrated by case 2, next-generation sequencing–based genetic testing is a powerful strategy for those cases with overlapping phenotypes or polygenic conditions.
Therapy of microcytic anemias: old and new approaches
In most of the hereditary microcytic anemias, the treatment is only supportive. Regarding the most common acquired IDA, oral iron therapy is the first choice for most of the patients.18 Numerous oral iron preparations exist, such as iron salts, ferrous sulfate, ferrous fumarate, and ferrous gluconate. If tolerated, this treatment should ameliorate the Hb levels within 2 to 3 weeks. An increase of Hb by 2 g/dL after 3 weeks of therapy is a commonly used criterion to define the patient’s hematological response to oral iron.19 For patients in whom oral iron therapy fails, the diagnosis of IRIDA becomes very suspicious, and, in such cases, IV iron is advisable. Several IV iron formulations are available and safe.20
On the contrary, for the iron overload microcytic anemias, the therapy aims to prevent the complications of iron accumulation in various organs and systems more than correcting the anemia, which can be mild/moderate. The treatment in such cases is based on iron chelation, and today there are 3 iron chelators in clinical practice: deferoxamine (due to the reduced half-life, it needs continuous subcutaneous infusion), deferiprone orally administered 3 times per day, and deferasirox that is orally administered in a single daily dose.12,20
Hepcidin, either in iron deficiency or in iron overload microcytic anemias, plays a major role; thus, currently, researchers are looking for new therapies able to modulate the hepcidin pathway. Indeed, hepcidin levels can help the response or resistance to oral iron administration, explaining part of iron refractoriness.21 Hepcidin agonists (that increase hepcidin levels), hepcidin antagonists (inhibitors of hepcidin synthesis), hepcidin binders (that block hepcidin function), and compounds that interfere with hepcidin–ferroportin interaction are in preclinical and clinical studies (Table 2).22,23 Hepcidin agonists can be used in anemias with ineffective erythropoiesis, such as β-thalassemia.24 The hepcidin analogs include minihepcidins, inhibitors of hepcidin repressors such as anti-TMPRSS6 molecules, and compounds that block ferroportin activity (Table 2). The use of these compounds demonstrated improvements in both anemia and iron overload in preclinical thalassemia models.25,26 The hepcidin agonists are currently used in phase I-II clinical trials. Moreover, drugs that improve erythroid maturation, such as the activin receptor IIB ligand trap, luspatercept, demonstrated their action not only by ameliorating anemia but also by reducing hepcidin inhibition.26 Hepcidin antagonists can be useful to release sequestered iron in IRIDA. In this latter condition, the use of a humanized antibody against hemojuvelin to modulate the hepcidin pathway has been proposed (Table 2).27
New drugs for the treatment of microcytic anemia
| Drug | Mechanism/effect | Preclinical data/clinical trial |
|---|---|---|
| Hepcidin analogs and mini-hepcidin | Replace endogenous hepcidin | Clinical trial |
| Anti-TMPRSS6 (ASO, siRNA) | Hepcidin inhibition | Clinical trial |
| FPN inhibitor VIT-2763 | Blocking the hepcidin receptor | Clinical trial |
| Transferrin injection | Decreasing transferrin receptor/reducing iron uptake | Preclinical study in animal model |
| Luspatercept | Activin receptor IIB ligand trap/ameliorating anemia and reducing hepcidin inhibition | Clinical trial |
| Protoporphyrin IX | Reducing iron cycling by inhibiting heme oxygenase 1 | Preclinical study in animal model |
| Anti–IL-6 and anti–IL-6R | Reducing the hepcidin signaling pathway | Preclinical study in animal model |
| Anti-BMP6 MoAb | Reducing the hepcidin signaling pathway | Clinical trial |
| BMP receptor inhibitors | Reducing the hepcidin signaling pathway | Preclinical study in animal model |
| Antihemojuvelin MoAb | Reduced hepcidin/correction of hypoferremia/correction of anemia | Preclinical study in animal model |
| Antihepcidin MoAb | Reduced hepcidin/correction of hypoferremia/correction of anemia | Preclinical study in animal model |
| Antihepcidin Spiegelmer | Reduced hepcidin/correction of hypoferremia/correction of anemia | Clinical trial |
| Antihepcidin anticalin | Reduced hepcidin/correction of hypoferremia/correction of anemia | Clinical trial |
| Antiferroportin MoAb GDP | Reduced hepcidin/correction of hypoferremia/correction of anemia | Preclinical study in animal model |
| Drug | Mechanism/effect | Preclinical data/clinical trial |
|---|---|---|
| Hepcidin analogs and mini-hepcidin | Replace endogenous hepcidin | Clinical trial |
| Anti-TMPRSS6 (ASO, siRNA) | Hepcidin inhibition | Clinical trial |
| FPN inhibitor VIT-2763 | Blocking the hepcidin receptor | Clinical trial |
| Transferrin injection | Decreasing transferrin receptor/reducing iron uptake | Preclinical study in animal model |
| Luspatercept | Activin receptor IIB ligand trap/ameliorating anemia and reducing hepcidin inhibition | Clinical trial |
| Protoporphyrin IX | Reducing iron cycling by inhibiting heme oxygenase 1 | Preclinical study in animal model |
| Anti–IL-6 and anti–IL-6R | Reducing the hepcidin signaling pathway | Preclinical study in animal model |
| Anti-BMP6 MoAb | Reducing the hepcidin signaling pathway | Clinical trial |
| BMP receptor inhibitors | Reducing the hepcidin signaling pathway | Preclinical study in animal model |
| Antihemojuvelin MoAb | Reduced hepcidin/correction of hypoferremia/correction of anemia | Preclinical study in animal model |
| Antihepcidin MoAb | Reduced hepcidin/correction of hypoferremia/correction of anemia | Preclinical study in animal model |
| Antihepcidin Spiegelmer | Reduced hepcidin/correction of hypoferremia/correction of anemia | Clinical trial |
| Antihepcidin anticalin | Reduced hepcidin/correction of hypoferremia/correction of anemia | Clinical trial |
| Antiferroportin MoAb GDP | Reduced hepcidin/correction of hypoferremia/correction of anemia | Preclinical study in animal model |
ASO, antisense specific oligonucleotides; BMP, bone morphogenetic protein; FPN, ferroportin; GDP, guanosine 5′-diphosphate encapsulated in lipid vesicle; IL, interleukin; MoAb, monoclonal antibodies; siRNA, short interfering RNA; VIT-2763, small molecule oral ferroportin inhibitor.
Correspondence
Maria Domenica Cappellini, Dipartimento di Scienze Cliniche e di Comunità, Università degli Studi di Milano, Via Luigi Anelli, 1, 20122 Milan, Italy; e-mail: maria.cappellini@unimi.it.
