

Abstract
Myelodysplastic syndrome (MDS)/myeloproliferative neoplasm (MPN) overlap syndromes are unique myeloid neoplasms, with overlapping features of MDS and MPN. They consist of four adult onset entities including chronic myelomonocytic leukemia (CMML), MDS/MPN-ring sideroblasts-thrombocytosis (MDS/MPN-RS-T), BCR-ABL1 negative atypical chronic myeloid leukemia (aCML) and MDS/MPN-unclassifiable (MDS/MPN-U); with juvenile myelomonocytic leukemia (JMML) being the only pediatric onset entity. Among these overlap neoplasms, CMML is the most frequent and is hallmarked by the presence of sustained peripheral blood monocytosis with recurrent mutations involving TET2 (60%), SRSF2 (50%) and ASXL1 (40%); with RAS pathway mutations and JAK2V617F being relatively enriched in proliferative CMML subtypes (WBC ≥13 × 109/L). CMML usually presents in the 7th decade of life, with a male preponderance and is associated with a median overall survival of <36 months. Adverse prognosticators in CMML include increasing age, high WBC, presence of circulating immature myeloid cells, anemia, thrombocytopenia and truncating ASXL1 mutations. While allogeneic stem cell transplantation remains the only curative option, given the late onset of this neoplasm and high frequency of comorbidities, most patients remain ineligible. Hypomethylating agents such as azacitidine, decitabine and oral decitabine/cedazuridine have been US FDA approved for the management of CMML, with overall response rates of 40-50% and complete remission rates of <20%. While these agents epigenetically restore hematopoiesis in a subset of responding patients, they do not impact mutational allele burdens and eventual disease progression to AML remains inevitable. Newer treatment modalities exploiting epigenetic, signaling and splicing abnormalities commonly seen in CMML are much needed.
Learning Objectives
Understand that chronic myelomonocytic leukemia (CMML) is an overlap syndrome that has features of myelodysplastic syndromes (anemia, thrombocytopenia, bone marrow dysplasia) and myeloproliferative neoplasms (leukocytosis, monocytosis, splenomegaly, and constitutional symptoms)
Understand that, although hypomethylating agents are able to epigenetically restore hematopoiesis in a subset of CMML patients, they fail to significantly alter mutational allele burdens and clonal transformation to acute myeloid leukemia
Clinical case
A 66-year-old male presents with progressive fatigue, unintentional weight loss, early satiety, and drenching night sweats. His complete blood count demonstrates a white blood cell (WBC) count of 44×109/L, 25% monocytes, circulating myelocytes/metamyelocytes, hemoglobin of 8.5 g/dL, and a platelet count of 110 × 109/L. On examination, he is found to have massive splenomegaly. A bone marrow (BM) biopsy was noted to be hypercellular (90%) and demonstrates megakaryocytic atypia, without megakaryocyte clusters and 10% BM blasts. The cytogenetics are normal (46, XY), BCR-ABL1 polymerase chain reaction testing negative, and molecular genetics identify mutations involving ASXL1, TET2, SRSF2, and NRAS. He is diagnosed as having chronic myelomonocytic leukemia-2 (CMML-2). His only associated comorbidity is hypertension, which is well controlled on lisinopril. His echocardiogram reveals a normal ejection fraction, and his hepatic, renal, and pulmonary function assessments are within normal limits. For this patient, would frontline therapy with hypomethylating agents (HMAs) or allogeneic stem cell transplantation (HCT) be preferred?
Introduction
Myelodysplastic syndrome (MDS)/myeloproliferative neoplasm (MPN) overlap syndromes are unique myeloid neoplasms with overlapping features of MDS and MPN. According to the 2016 World Health Organization (WHO) classification, MDS/MPN overlap neoplasms consist of 4 adult-onset entities, including CMML, BCR-ABL1− atypical chronic myeloid leukemia (aCML), MDS/MPN-ring sideroblasts and thrombocytosis (MDS/MPN-RS-T), and MDS/MPN-unclassifiable (MDS/MPN-U), along with 1 pediatric entity, juvenile myelomonocytic leukemia (JMML) (Table 1).1 Among these, CMML is the most common (crude and age-standardized incidence ratios per 100 000 people in the United States are as follows: CMML, 0.6 (0.57-0.63); aCML, 0.06 (0.04-0.62); MDS/MPN-U, 0.07 (0.006-0.009); MDS/MPN-RS-T < 1% of new MDS cases; and JMML, 1.2 per 1 000 000 people)2 and is a clonal stem cell disorder that is characterized by sustained peripheral blood (PB) monocytosis (≥1 × 109/L and ≥10% of WBC differential) and an inherent tendency for transformation to acute myeloid leukemia (AML; 15-20% over 3-5 years).3 The median age at diagnosis for CMML is 73 years, with a male preponderance.3 Histologically, CMML can be classified as CMML-0 (<2% PB blasts and <5% bone marrow [BM] blasts), CMML-1 (2-4% PB blasts and/or 5-9% BM blasts), and CMML-2 (5-19% PB blasts and/or 10-19% BM blasts or when Auer rods are present). Clinically, and based on the presenting WBC count, CMML can be classified as MPN-CMML (WBC count ≥ 13 × 109/L) or MDS-CMML (WBC count < 13 × 109/L); the former has poor outcomes and higher rates of AML transformation.1,3
2016 WHO diagnostic criteria for MDS/MPN overlap neoplasms
| MPN/MDS overlap subtypes | 2016, WHO diagnostic criteria* | Frequency of somatic gene mutations | Median age at Dx, y | Median OS | Rate of LT |
|---|---|---|---|---|---|
| aCML | WBC count > 13 × l09/L with increased and dysplastic neutrophils. No or minimal absolute basophils and monocytosis. Hypercellular BM with granulocytic proliferation and dysplasia (neutrophil precursors greater than equal to 10%). | ASXL1 (60%), SETBP1 (48%), N/KRAS (35%), TET2 (30%), EZH2 (13%) and <10% for the following: CSF3R, ETNK1, CBL, FLT3, RUNX1, CEBPA, IDH1/2 | >60 | 22 mo | 30-40% |
| CMML | Persistent PB monocytosis ≥ 1 × 109/L. Dysplasia in ≥1 lineage, if no dysplasia, then must include an acquired clonal cytogenetic or molecular genetic abnormality (TET2, ASXL1, SRSF2, and/or SETBP1). | TET2 (60%), SRSF2 (50%), ASXL1 (40%), NRAS (15%), CBL (15%), RUNX1 (15%), SETBP1 (15%), KRAS (10%), IDH1/2 (5-10%), and <10% for the following: JAK2, SF3B1, U2AF1, EZH2, DNMT3A, PTPN11, ZRSR2, FLT3 | 71-74 | 28-32 mo | 15-30% |
| MDS/MPN-RS-T | Platelet count ≥ 450 × 109/L. 15% ring sideroblasts in the BM or >5% with SF3B1 mutation. Presence of megakaryocytic atypia resembling ET or MF. | SF3B1 (93%), JAK2 (57%), TET2 (25%), DNMT3A (15%), ASXL1 (15%), and <10% for the following: SRSF2, CBL, SETBP1, IDHl/2 | 71-75 | 76 mo | 1-2% |
| MDS/MPN-U | Myeloid neoplasm with mixed MDS and MPN features, not meeting WHO criteria for other MDS/MPN overlap neoplasms, MDS or MPN. | TET2 (30%), RUNX1 (14%), CBL (11%), EZH2 (10%), N/KRAS (10%), SETBP1 (10%), and <10% for the following: DNMT3A, CEBPA, IDH1/2 | 70 | 12-28 mo | Unknown |
| JMML | PB monocyte count ≥ 1 × 109/L. Splenomegaly. Genetic features (must include 1 of the following): somatic mutation in PTPN11,† KRAS,† or NRAS†; diagnosis of neurofibroatosis-1 or NF1 mutation; or germline CBL mutation and loss of heterozygosity of CBL. If no genetic features then, must have a clonal chromosomal abnormality or all of the following: GM-CSF hypersensitivity, hyperphosphorylation of STAT5, fetal hemoglobin increased for age, myeloid or erythroid precursors on PB smear | PTPN11† (38%), NRAS† (18%), KRAS† (14%), CBL (12-18%), NF1 (5-10%) | 1.4-2 | 10-12 mo | Infrequent |
| MPN/MDS overlap subtypes | 2016, WHO diagnostic criteria* | Frequency of somatic gene mutations | Median age at Dx, y | Median OS | Rate of LT |
|---|---|---|---|---|---|
| aCML | WBC count > 13 × l09/L with increased and dysplastic neutrophils. No or minimal absolute basophils and monocytosis. Hypercellular BM with granulocytic proliferation and dysplasia (neutrophil precursors greater than equal to 10%). | ASXL1 (60%), SETBP1 (48%), N/KRAS (35%), TET2 (30%), EZH2 (13%) and <10% for the following: CSF3R, ETNK1, CBL, FLT3, RUNX1, CEBPA, IDH1/2 | >60 | 22 mo | 30-40% |
| CMML | Persistent PB monocytosis ≥ 1 × 109/L. Dysplasia in ≥1 lineage, if no dysplasia, then must include an acquired clonal cytogenetic or molecular genetic abnormality (TET2, ASXL1, SRSF2, and/or SETBP1). | TET2 (60%), SRSF2 (50%), ASXL1 (40%), NRAS (15%), CBL (15%), RUNX1 (15%), SETBP1 (15%), KRAS (10%), IDH1/2 (5-10%), and <10% for the following: JAK2, SF3B1, U2AF1, EZH2, DNMT3A, PTPN11, ZRSR2, FLT3 | 71-74 | 28-32 mo | 15-30% |
| MDS/MPN-RS-T | Platelet count ≥ 450 × 109/L. 15% ring sideroblasts in the BM or >5% with SF3B1 mutation. Presence of megakaryocytic atypia resembling ET or MF. | SF3B1 (93%), JAK2 (57%), TET2 (25%), DNMT3A (15%), ASXL1 (15%), and <10% for the following: SRSF2, CBL, SETBP1, IDHl/2 | 71-75 | 76 mo | 1-2% |
| MDS/MPN-U | Myeloid neoplasm with mixed MDS and MPN features, not meeting WHO criteria for other MDS/MPN overlap neoplasms, MDS or MPN. | TET2 (30%), RUNX1 (14%), CBL (11%), EZH2 (10%), N/KRAS (10%), SETBP1 (10%), and <10% for the following: DNMT3A, CEBPA, IDH1/2 | 70 | 12-28 mo | Unknown |
| JMML | PB monocyte count ≥ 1 × 109/L. Splenomegaly. Genetic features (must include 1 of the following): somatic mutation in PTPN11,† KRAS,† or NRAS†; diagnosis of neurofibroatosis-1 or NF1 mutation; or germline CBL mutation and loss of heterozygosity of CBL. If no genetic features then, must have a clonal chromosomal abnormality or all of the following: GM-CSF hypersensitivity, hyperphosphorylation of STAT5, fetal hemoglobin increased for age, myeloid or erythroid precursors on PB smear | PTPN11† (38%), NRAS† (18%), KRAS† (14%), CBL (12-18%), NF1 (5-10%) | 1.4-2 | 10-12 mo | Infrequent |
Dx, diagnosis; ET, essential thrombocythemia; GM-CSF, granulocyte-macrophage colony-stimulating factor; LT, Leukemic transformation; MF, myelofibrosis; OS, overall survival; STAT5- signal tansducer and activator of transcription 5.
All MDS/MPN overlap subtypes are negative for BCR-ABL1 fusions, or rearrangements involving PDGFRA, PDGFRB, FGFR1 and PCM1-JAK2 and have <20% blasts in the PB and BM.1
†Germline mutations (indicative of Noonan syndrome) need to be excluded.
Diagnosis
CMML is diagnosed based on the presence of sustained PB monocytosis (>3 months and in the absence of reactive etiologies), the absence of molecular aberrations that can be associated with monocytosis (PDGFRA, PDGFRB, PCM1-JAK2, and BCR-ABL1), and <20% PB and BM blasts, with or without BM dysplasia.1,3 In the absence of BM dysplasia, WHO criteria allow ascertainment of clonality by demonstration of clonal cytogenetic abnormalities or gene mutations involving TET2, ASXL1, SRSF2, and SETBP1.1,3 Recently, PB flow cytometry demonstrating monocyte subset repartitioning has become a useful adjunct in CMML diagnosis, with most CMML patients demonstrating an expansion (>94%) of classical monocytes (MO1−CD14+/CD16−). This modality has also shown promise in distinguishing CMML from other causes of monocytosis, including MPN with monocytosis. CMML patients usually have between 10 and 12 mutations per kilobase of DNA coding region, with the 3 most frequently mutated genes being TET2 (60%), SRSF2 (50%), and ASXL1 (40%).4 Cytogenetic abnormalities are seen in 20% to 30% of patients, with no abnormality being unique to CMML. Importantly, given the high frequency of TET2 mutations in CMML and the reported convergence of mutant TET2 and IDH pathways (2-hydroxyglutarate–mediated suppression of TET2 activity), IDH1/2 mutations are infrequent in CMML (<10%).4
Pathobiology
CMML is a disease of ageing, with CMML-related driver mutations occurring in the context of age-related clonal hematopoiesis.5 Mutations involving TET2 (epigenetic) and SRSF2 (splicing) often skew hematopoiesis toward monocytosis, with subsequent mutations in epigenetic regulators (ASXL1) or signal pathways (NRAS, CBL, KRAS, PTPN11, and JAK2) giving rise to disease. MPN-CMML is enriched in active RAS/MAPK signaling, with ∼70% of patients demonstrating RAS pathway mutations; NRAS is the most common. RAS pathway mutations, along with epigenetic events, also play a role in CMML transformation to AML. The classical monocytes expanded in CMML produce an inflammatory milieu, resulting in elevated levels of tumor necrosis factor-α, interleukin-6, and interleukin-8, which can result in exaggerated leukemoid reactions and cytokine release syndromes.
Prognosis
Several prognostic models exist for CMML, with 3 contemporary models integrating the aforementioned molecular abnormalities: the Mayo molecular model (ASXL1), Groupe Francophone des Myélodysplasies model (ASXL1), and the CMML-specific prognostic scoring system-molecular model (ASXL1, RUNX1, NRAS, SETBP1 and cytogenetic abnormalities) (Table 2).3,6 Frameshift and nonsense mutations in ASXL1 truncate the protein and are universally deleterious in CMML, with the ASXL1wt/TET2mt genotype having the best outcomes, including response to HMA therapy.4 Clinical factors adversely impacting outcomes include anemia, advanced age, leukocytosis, monocytosis, circulating immature myeloid cells (IMCs), and thrombocytopenia.3
Molecularly integrated CMML prognostic Scoring Systems
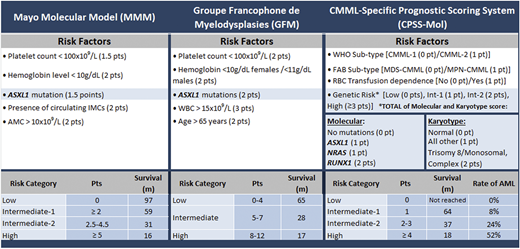
AMC, absolute monocyte count; FAB, French-American-British; Int-1, intermediate-1; Int-2, intermediate-2; m, month; MDS-CMML, myelodysplastic syndrome-like CMML; MPN-CMML, myeloproliferative neoplasm-like CMML; pt/pts/Pts, point(s); RBC, red blood cell; rate of AML, rate of transformation (cumulative at 48 mo).
Management
Allogeneic HCT remains the only curative modality; however, eligibility includes <10% of patients because of their advanced age at presentation and comorbidities. HCT is also fraught with morbidity (eg, graft-versus-host disease) and mortality and tends to be poorly tolerated by older frail patients. However, the advent of reduced-intensity conditioning (RIC) and the use of alternate donor sources continue to expand eligibility and improve tolerability. In general, HCT outcomes have been suboptimal, with 5-year overall survival rates of 30% to 40% and transplant-related mortality rates of 20% to 30%.3 In a large registry study (Center for International Blood and Marrow Transplant Research, N = 209), overall survival rates at 1, 3, and 5 years were 61%, 48%, and 41%, respectively, for CPSS low/intermediate-1–risk patients and were 38%, 32%, and 19%, respectively, for intermediate-2/high–risk patients.7 In younger patients, retrospective studies have shown better outcomes, in particular with myeloablative conditioning.8 Nevertheless, given the absence of prospective randomized studies, questions with regard to optimal timing for HCT in CMML, disease risk–based selection criteria (lower vs higher risk), and the role for pre-HCT therapies (HMAs) remain to be answered. In general, extrapolating from data available in patients with MDS and MPN, HCT in CMML is reserved for higher-risk patients with acceptable organ functions and appropriately matched donor options.
For HCT-ineligible patients, in addition to supportive care measures, HMAs (5-azacitidine and decitabine) are often used to manage disease-related features.3 These drugs have been approved based on the inclusion of a small number of predominantly MDS CMML patients in MDS-predominant phase 3 clinical trials; there has not been any prospective phase 3 randomized study proving their efficacy in CMML. In general, HMAs act by inhibiting DNA methyltransferases, reducing genome-wide and sequence-specific methylation changes that contribute to neoplasia (Figure 1). HMAs are associated with an overall response rate of 40% to 50%, with true complete remission rates < 20%; with previously responding patients on therapy demonstrating AML transformation.9 Elaborate sequencing studies have demonstrated that, although HMAs can epigenetically restore normal hematopoiesis in a subset of CMML patients, they fail to alter mutational allele burdens, even in responders, with disease evolution being inevitable in the vast majority (Figure 1).10 In addition, smaller prospective studies and retrospective data have revealed that these drugs are particularly ineffective in MPN-CMML subtypes.11 That being said, epigenetic restoration of hematopoiesis is useful in alleviating cytopenias and cytopenia-related symptoms, as well as in reducing the need for red blood cell and platelet transfusions. In addition, HMAs have a direct cytotoxic effect and can be used to reduce PB and BM blasts in patients with advanced disease.
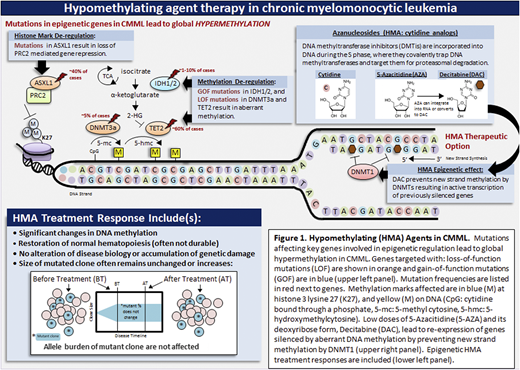
HMAs in CMML. Mutations affecting key genes involved in epigenetic regulation lead to global hypermethylation in CMML. Genes targeted with loss-of-function (LOF) mutations are shown in orange and gain-of-function mutations (GOF) are in blue (upper left panel). Mutation frequencies are listed in red next to genes. Methylation marks affected are in blue (M) at histone 3 lysine 27 (K27) and yellow (M) on DNA. Low doses of 5-azacitidine and its deoxyribose form, decitabine, lead to re-expression of genes silenced by aberrant DNA methylation by preventing new strand methylation by DNA methyltransferases (upper right panel). Epigenetic HMA treatment responses are included (lower left panel). CpG, cytidine bound through a phosphate; 5-hmc, 5-hydroxymethylcytosine; 5-mc, 5-methylcytosine.
HMAs in CMML. Mutations affecting key genes involved in epigenetic regulation lead to global hypermethylation in CMML. Genes targeted with loss-of-function (LOF) mutations are shown in orange and gain-of-function mutations (GOF) are in blue (upper left panel). Mutation frequencies are listed in red next to genes. Methylation marks affected are in blue (M) at histone 3 lysine 27 (K27) and yellow (M) on DNA. Low doses of 5-azacitidine and its deoxyribose form, decitabine, lead to re-expression of genes silenced by aberrant DNA methylation by preventing new strand methylation by DNA methyltransferases (upper right panel). Epigenetic HMA treatment responses are included (lower left panel). CpG, cytidine bound through a phosphate; 5-hmc, 5-hydroxymethylcytosine; 5-mc, 5-methylcytosine.
The current patient is a 66-year-old male with high-risk CMML based on his BM blast percentage and the presence of high-risk features, including leukocytosis, monocytosis, circulating IMCs, anemia, thrombocytopenia, and the presence of ASXL1 and NRAS mutations. Given his age, he is not a candidate for a myeloablative conditioning HCT and would need an RIC regimen, once an appropriate donor is identified. His HCT comorbidity index is acceptable (http://www.hctci.org/Home/Calculator); given the higher relapse rates with RIC HCT, he will benefit from disease control prior to HCT. In his case, HMA therapy with 4 to 6 cycles of 5-azacitidine was recommended, with the goal of reducing BM blasts to <5% and then proceeding with an RIC allogeneic HCT with an appropriately matched donor.
In summary, although advances in CMML biology have clearly been made, therapeutic options for affected patients remain limited. Although HMAs do have a role to play in ameliorating cytopenias and possibly as a bridge to allogeneic HCT, they do not alter mutational allele burdens, and disease progression remains inevitable. In addition, their efficacy in MPN-CMML subtypes is limited. Better elucidation of genetic (RAS signaling and splicing) and epigenetic (TET2 and ASXL1) events frequently seen in CMML and the development of rationally derived therapies targeting exposed vulnerabilities are much needed.
Acknowledgment
This work was supported by a grant from the Henry J. Predolin Foundation for Research in Leukemia (Mayo Clinic, Rochester, MN).
Correspondence
Mrinal M. Patnaik, Division of Hematology, Mayo Clinic, 200 First St SW, Rochester, MN 55901; e-mail: patnaik.mrinal@mayo.edu.
