
Abstract
Acute lymphoblastic leukemia (ALL) is characterized by genetic alterations that block differentiation, promote proliferation of lymphoid precursor cells, and are important for risk stratification. Although ALL is less common in adolescents and young adults (AYAs) and adults than children, survival rates are inferior, and long-term prognosis for adults is poor. Thus, ALL remains a challenging disease to treat in the AYA and adult populations. A major contributing factor that influences prognosis in this population is the reduced prevalence of genetic subtypes associated with favorable outcome and a concomitant increase in subtypes associated with poor outcome. Recent advances in genomic profiling across the age spectrum continue to enhance our knowledge of the differences in disease biology between children and adults and are providing important insights into novel therapeutic targets. Philadelphia chromosome-like (Ph-like) ALL is one such subtype characterized by alterations that deregulate cytokine receptor or tyrosine kinase signaling and are amenable to inhibition with approved tyrosine kinase inhibitors. One of the greatest challenges now remaining is determining how to implement this breadth of genomic information into rapid and accurate diagnostic testing to facilitate the development of novel clinical trials that improve the outcome of AYAs and adults with ALL.
Learning Objectives
Review new genetic subtypes identified in acute lymphoblastic leukemia (ALL)
Understand the biological differences between children and adults
Discuss the implementation of genomic information in the clinic for improved management of ALL
Introduction
B-precursor acute lymphoblastic leukemia (B-ALL) comprises multiple genetic subtypes characterized by founding chromosomal alterations that are important for risk stratification, including aneuploidy (gains and losses of whole chromosomes), or chromosomal rearrangements that result in deregulation of genes by juxtaposition to strong enhancers or the formation of chimeric fusion genes, commonly involving hematopoietic transcription factors, epigenetic modifiers, cytokine receptors, and tyrosine kinases (Table 1).1 Secondary genomic events that contribute to leukemogenesis include copy number alterations (commonly involving lymphoid transcription factors) and sequence mutations. Children (age younger than 15 years) diagnosed with acute lymphoblastic leukemia (ALL) have an excellent prognosis, with cure rates exceeding 85%.2 However, the prognosis for ALL declines with increasing age, with historic cure rates of just 30% to 40% in adults (age older than or equal to 40 years),3 and relapsed ALL remains a major cause of cancer-related death for all ages. Data from the US Surveillance, Epidemiology and End Results (SEER) database from all patients with ALL diagnosed between 2000 and 2007 show that the survival of ALL is triphasic, with survival rates of 75% at 17 years, 48% at 20 years, and 15% at 70 years. The significant drop from ages 17 to 20 years accounts for 45% of the total survival decrease between the ages of 17 to 70 years and is known as the “survival cliff.”4 Certainly, the outcome of adolescents and young adults (AYAs) with ALL (15-39 years) has improved significantly in the last decade due to the increased use of pediatric-inspired regimens.4 Stock et al5 report event-free survival and overall survival rates of 66% and 79% from the C10403 trial that treated 296 evaluable patients ages 17 to 39 years. However, access to clinical trials and pediatric regimens is not readily available to all AYA patients.6
Prevalence and prognosis of subtypes in Ba-ALL
| ALL subtype | Category | Median age, y | Prevalence | Genomic alterations | Clinical features | References | |
|---|---|---|---|---|---|---|---|
| Hyperdiploid (>50 chromosomes) | Aneuploid | 4 | High in children (25%) | Ras pathway, epigenetic modifiers | Excellent prognosis | 9 | |
| Low hypodiploid (31-39 chromosomes) | Aneuploid | 47 | High in adults (10%-15%) | IKZF2 del, TP53 mut (commonly inherited) | Poor prognosis | 10 | |
| Near haploid (24-30 chromosomes) | Aneuploid | 5 | <3% in all ages | Ras pathway, IKZF3 del | Intermediate prognosis | 10 | |
| iAMP21 | Copy number gain | 10 | ∼3% in children and AYAs | Complex structural alterations of chromosome 21 | Good prognosis with intensive therapy, low WBC | 12 | |
| ETV6-RUNX1 t(12;21)(p13;q22) | TF rearrangement | 4 | High in children (25%) | PAX5 del, WHSC1 mut | Excellent prognosis | 8,13 | |
| ETV6-RUNX1 like | TF rearrangement | 3 | ∼3% in children | ETV6 fusions and del, IKZF1 fusions and del | Unknown | 34,35 | |
| DUX4 rearranged | TF rearrangement | 14 | Peak in AYAs (∼8%) | ERG del, IKZF1 del, Ras pathway | Excellent prognosis | 34,36,37 | |
| KMT2A rearranged | TF rearrangement | 40 | High in infants (∼90%) and adults (∼15%) | Ras pathway (commonly subclonal) | Poor prognosis, sensitive to bortezomib or DOT1L inhibition | 14 | |
| TCF3-PBX1 t(1;19)(q23;p13) | TF rearrangement | 8 | ∼5% in children, rarely in adults | Good prognosis, CNS relapse | 15,16 | ||
| ZNF384 rearranged | TF rearrangement | 15 | Peak in AYAs (∼5%) | Epigenetic modifiers, Ras pathway | Intermediate prognosis | 36,41,42 | |
| MEF2D rearranged | TF rearrangement | 14 | Peak in AYAs (∼7%) | Ras pathway | Intermediate prognosis, sensitive to HDAC inhibition | 39,40 | |
| NUTM1 rearranged | TF rearrangement | 3 | Exclusively in children (1%) | Unknown | Excellent prognosis | ||
| TCF3-HLF t(17;19)(q22;p13) | TF rearrangement | 15 | Very rare in all ages (<1%) | TCF3 mut, PAX5 del, Ras pathway | Very poor prognosis, sensitive to Bcl2 inhibition | 17 | |
| PAX5alt | Other TF driven | 10 | Highest in children (∼11%) | PAX5 fusion, mut, amp | Intermediate prognosis | ||
| PAX5 P80R | Other TF driven | 22 | Highest in adults (∼4%) | Signaling alterations | Unclear | ||
| BCL2/MYC rearranged | Other TF driven | 48 | Almost exclusively in AYAs and adults (∼3%) | Unknown | Poor prognosis | ||
| Ph-like | Kinase driven | 21 | Peaks in AYAs (25%-30%) | Multiple kinase alterations, IKZF1 del and mut, CDKN2A/B del | Poor prognosis, amenable to TKI therapy | 18,19 | |
| BCR-ABL1 t(9;22)(q34;q11.2) | Kinase driven | 40-45 | 5% in children; highest in adults (40%-50%) | IKZF1 del and mut, CDKN2A/B del | Historically poor prognosis, improved with TKI | 18,19,23 | |
| Other | 16 | ∼5% children; ∼10% AYAs and adults | Unknown | Intermediate prognosis |
| ALL subtype | Category | Median age, y | Prevalence | Genomic alterations | Clinical features | References | |
|---|---|---|---|---|---|---|---|
| Hyperdiploid (>50 chromosomes) | Aneuploid | 4 | High in children (25%) | Ras pathway, epigenetic modifiers | Excellent prognosis | 9 | |
| Low hypodiploid (31-39 chromosomes) | Aneuploid | 47 | High in adults (10%-15%) | IKZF2 del, TP53 mut (commonly inherited) | Poor prognosis | 10 | |
| Near haploid (24-30 chromosomes) | Aneuploid | 5 | <3% in all ages | Ras pathway, IKZF3 del | Intermediate prognosis | 10 | |
| iAMP21 | Copy number gain | 10 | ∼3% in children and AYAs | Complex structural alterations of chromosome 21 | Good prognosis with intensive therapy, low WBC | 12 | |
| ETV6-RUNX1 t(12;21)(p13;q22) | TF rearrangement | 4 | High in children (25%) | PAX5 del, WHSC1 mut | Excellent prognosis | 8,13 | |
| ETV6-RUNX1 like | TF rearrangement | 3 | ∼3% in children | ETV6 fusions and del, IKZF1 fusions and del | Unknown | 34,35 | |
| DUX4 rearranged | TF rearrangement | 14 | Peak in AYAs (∼8%) | ERG del, IKZF1 del, Ras pathway | Excellent prognosis | 34,36,37 | |
| KMT2A rearranged | TF rearrangement | 40 | High in infants (∼90%) and adults (∼15%) | Ras pathway (commonly subclonal) | Poor prognosis, sensitive to bortezomib or DOT1L inhibition | 14 | |
| TCF3-PBX1 t(1;19)(q23;p13) | TF rearrangement | 8 | ∼5% in children, rarely in adults | Good prognosis, CNS relapse | 15,16 | ||
| ZNF384 rearranged | TF rearrangement | 15 | Peak in AYAs (∼5%) | Epigenetic modifiers, Ras pathway | Intermediate prognosis | 36,41,42 | |
| MEF2D rearranged | TF rearrangement | 14 | Peak in AYAs (∼7%) | Ras pathway | Intermediate prognosis, sensitive to HDAC inhibition | 39,40 | |
| NUTM1 rearranged | TF rearrangement | 3 | Exclusively in children (1%) | Unknown | Excellent prognosis | ||
| TCF3-HLF t(17;19)(q22;p13) | TF rearrangement | 15 | Very rare in all ages (<1%) | TCF3 mut, PAX5 del, Ras pathway | Very poor prognosis, sensitive to Bcl2 inhibition | 17 | |
| PAX5alt | Other TF driven | 10 | Highest in children (∼11%) | PAX5 fusion, mut, amp | Intermediate prognosis | ||
| PAX5 P80R | Other TF driven | 22 | Highest in adults (∼4%) | Signaling alterations | Unclear | ||
| BCL2/MYC rearranged | Other TF driven | 48 | Almost exclusively in AYAs and adults (∼3%) | Unknown | Poor prognosis | ||
| Ph-like | Kinase driven | 21 | Peaks in AYAs (25%-30%) | Multiple kinase alterations, IKZF1 del and mut, CDKN2A/B del | Poor prognosis, amenable to TKI therapy | 18,19 | |
| BCR-ABL1 t(9;22)(q34;q11.2) | Kinase driven | 40-45 | 5% in children; highest in adults (40%-50%) | IKZF1 del and mut, CDKN2A/B del | Historically poor prognosis, improved with TKI | 18,19,23 | |
| Other | 16 | ∼5% children; ∼10% AYAs and adults | Unknown | Intermediate prognosis |
ALL, acute lymphoblastic leukemia; amp, amplification; AYA, adolescent and young adult; CNS, central nervous system; del, deletion; DUX4, double homeobox 4; ERG, ETS transcription factor; HDAC, histone deacetylase; iAMP21, intrachromosomal amplification of chromosome 21; MEF2D, myocyte enhancer factor 2D; mut, sequence mutation; NUTM1, nuclear protein in testis midline carcinoma family 1; Ph like, Philadelphia chromosome like; TF, transcription factor; TKI, tyrosine kinase inhibitor; WBC, white blood cell; ZNF384, zinc finger 384.
The age-related decline in survival observed in the SEER database is partly explained by a reduced prevalence of genetic alterations associated with favorable outcome (eg, ETV6-RUNX1) and a concurrent increase in genetic alterations associated with poor outcome (eg, BCR-ABL1). In recent years, significant progress has been gained in treating AYAs due to several factors including but not limited to (1) recognition of this group as a unique population, (2) development of treatment protocols (particularly pediatric-inspired regimens) specifically designed for AYAs and increasing the availability of clinical trials, and (3) improvements in our understanding of the biological differences in ALL across the age spectrum.4 This review will outline the genomic landscape of B-ALL with particular emphasis on new subtypes and prognosis and discuss the biological differences between children and adults. The role and implementation of genomic testing to improve the clinical management of ALL will also be discussed.
Recurring chromosomal alterations and prognosis
Recurring chromosomal rearrangements are a hallmark of ALL, and the frequency of each varies with age (Figure 1; Table 1). Secondary genetic lesions, including copy number alterations (loss and gain of DNA) and sequence mutations that perturb key cellular pathways, are important cooperating lesions in leukemogenesis and may be acquired or enriched during disease progression.7,8 Common targets include lymphoid transcription factors (IKZF1, PAX5, EBF1, and ETV6), cell cycle regulators and tumor suppressors (CDKN2A/B, TP53, and RB1), regulators of lymphoid signaling (BTLA and CD200), and chromatin modifiers (CREBBP, SETD2, and WHSC1).8 The prevalence, gene, and type of alteration vary between subtypes and have different prognostic relevance.
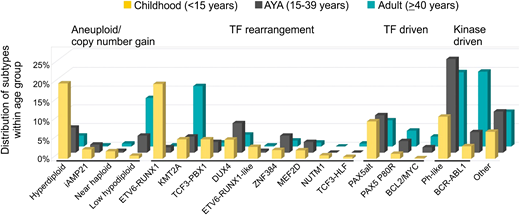
Distribution of B-ALL subtypes within each age group. Subtypes are grouped as aneuploid/copy number gain, transcription factor (TF) rearrangement, other TF driven, kinase driven, and all others.
Distribution of B-ALL subtypes within each age group. Subtypes are grouped as aneuploid/copy number gain, transcription factor (TF) rearrangement, other TF driven, kinase driven, and all others.
Aneuploidy and copy number gain
High hyperdiploidy (nonrandom gain of at least 5 chromosomes) is present in ∼25% of childhood ALL patients but accounts for <5% of AYAs and adults, and it is associated with a favorable outcome. Mutations involving the Ras pathway (KRAS, NRAS, and PTPN11) and epigenetic modifiers are frequent genetic events in hyperdiploidy patients.9 Hypodiploid ALL with <44 chromosomes comprises 2 subtypes with distinct transcriptional profiles and genetic alterations. Low-hypodiploidy patients (31-39 chromosomes) commonly harbor deletion of IKZF2 and sequence mutations of TP53 that are frequently inherited.10 This subtype is extremely rare in children (<1%) but increases significantly with age, accounting for 5% of AYAs and over 10% of adults, and it is associated with a very poor outcome.11 Near-haploid ALL (24-30 chromosomes) is characterized by Ras-activating mutations and IKZF3 alterations, accounting for ∼2% of childhood ALL and <1% of AYAs and adults.10 Doubling of the hypodiploid clone (known as masked hypodiploidy) is common in both near-haploid and low-hypodiploid ALL and results in a modal chromosome number in the hyperdiploid range. Accurate classification is important for risk stratification, because masked hypodiploidy can be confused with hyperdiploidy with an opposite good prognosis. Intrachromosomal amplification of chromosome 21 is more common in older children (median age of 10 years), and it is rarely observed in patients older than 30 years. Improved risk stratification and treatment with intensive therapy can rescue the poor outcome of these patients when treated as standard risk.12
Translocations
The ETV6-RUNX1 gene fusion, encoded by the t(12;21)(p13;q22) translocation, is another known favorable cytogenetic alteration with a high frequency in childhood ALL (25%) and <5% in AYAs and adults. PAX5 deletion and mutation of WHSC1 are frequent in ETV6-RUNX1–positive ALL.8,13 The age distribution of KMT2A rearrangements (11q23) in ALL is biphasic. Their presence is a hallmark of infant ALL (age younger than 1 year), with increased prevalence observed in AYAs (4%) and peaking again in adults (∼15%). The mechanism underlying these 2 peaks in age is unclear. KMT2A rearrangements are associated with a poor prognosis2 and very few secondary alterations, suggesting that the rearrangement itself is sufficient to induce leukemia.14 TCF3-PBX1, encoded by the t(1;19)(q23;p13) translocation, is present in ∼5% of children and fewer AYAs and adults. Previously considered a high-risk subtype, it is now associated with a favorable outcome on contemporary ALL therapies.15,16 By contrast, the t(17;19)(q22;p13) translocation, encoding the TCF3-HLF fusion gene, defines a rare subtype of ALL (<1% in all ages) that is typically associated with relapse and death within 2 years from diagnosis. Interestingly, primary leukemic cells harboring TCF3-HLF show sensitivity to the Bcl2 inhibitor, venetoclax (ABT-199), identifying a new therapeutic option for this fatal subtype.17 BCR-ABL1 ALL is uncommon in children (2%-5% of patients) but accounts for 6% of AYAs and at least 25% of adults.18,19 The survival of patients with BCR-ABL1 has been markedly improved with the use of tyrosine kinase inhibitors (TKIs) in both children and adults.20-22 IKZF1 alterations are a hallmark of kinase-driven ALL (BCR-ABL1 and Philadelphia chromosome like [Ph like]), and they are associated with treatment failure and relapse.19,23 The co-occurrence of IKZF1 deletions with CDKN2A/B, PAX5, or PAR1 deletions (termed IKZF1plus) in childhood ALL confers a worse prognosis compared with patients with IKZF1 deletion who do not fulfill the criteria for IKZF1plus.24 A recent study by Mullighan and coworkers reported inherited germline variants in IKZF1 that impair its function in a similar manner to somatic mutations by conferring stem cell–like properties and increased adhesion, further highlighting the importance of this gene in both de novo and familial ALL.25,26
New subtypes in B-ALL
The advent of next generation sequencing and comprehensive integrative analyses has rapidly increased our understanding of the genomic landscape of ALL, resulting in the identification of new subtypes with prognostic and therapeutic significance. In contrast to subtypes characterized by aneuploidy or a single chromosomal rearrangement (eg, ETV6-RUNX1 or BCR-ABL1), rearrangements in these new subtypes are commonly cryptic by cytogenetic analysis (double homeobox 4 [DUX4]-rearranged ALL) or involve a diverse range of partners that converge on a single gene (myocyte enhancer factor 2D [MEF2D] and zinc finger 384 [ZNF384]-rearranged ALL). Additional groups are phenocopies of known subtypes with diverse genetic alterations (Ph-like and ETV6-RUNX1–like ALL).
Ph-like ALL: opportunity for targeted therapies
A major advance in understanding the differences in outcome between children and AYA/adults has been the identification of a high-risk subtype termed Ph-like (BCR-ABL1–like) ALL. This subtype has a gene expression signature similar to Ph-positive ALL but lacks the BCR-ABL1 fusion gene.27,28 The 2016 revision to the World Health Organization classification of acute leukemia incorporated Ph-like ALL as a provisional entity into the classification of B-ALL.29
Ph-like ALL is a heterogeneous subtype characterized by rearrangements, copy number alterations, and sequence mutations that activate tyrosine kinase or cytokine receptor signaling. Despite this complexity, a majority of alterations can be divided into a limited number of distinct subgroups based on the activated kinase and signaling pathways. These include rearrangements of CRLF2 (IGH-CRLF2 and P2RY8-CRLF2), fusions involving ABL-class genes (ABL1, ABL2, CSF1R, LYN, PDGFRA, and PDGFRB), rearrangements of JAK2 or EPOR, alterations activating JAK-STAT (IL7R, SH2B3, JAK1, JAK3, TYK2, and IL2RB) or Ras signaling pathways (NRAS, KRAS, and PTPN11), and other less common fusions (FLT3, FGFR1, and NTRK3).18,19,30 The frequency of each kinase subgroup varies with age, particularly with respect to CRLF2 rearrangements, where IGH-CRLF2 accounts for almost 50% of Ph-like ALL in AYAs and adults. ABL-class fusions are most prevalent in children with National Cancer Institute (NCI) high-risk ALL. Fewer kinase alterations are identified in Ph-like ALL patients with NCI standard-risk ALL31 (Figure 2).
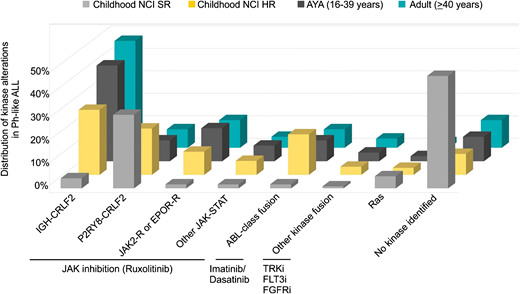
Distribution of kinase subtypes in Ph-like ALL within each age group.18,19,30,31 Combined prevalence of Ph-like ALL subtypes in childhood NCI standard risk (SR; age 1-9.99 years and white blood cells <50 000/μL), NCI high risk (HR; age 10-15 years or white blood cells ≥50 000/μL), AYAs (16-39 years), and adults (older than or equal to 40 years). Genomic subtypes include IGH-CRLF2, P2RY8-CRLF2, ABL-class fusions (ABL1, ABL2, CSF1R, LYN, PDGFRA, and PDGFRB), JAK2 and EPOR rearrangements, other mutations in JAK-STAT signaling (JAK1/3, IL7R, SH2B3, TYK2, and IL2RB), other kinase alterations (FLT3, FGFR1, and NTRK3,), Ras mutations (KRAS, NRAS, NF1, PTPN11, BRAF, and CBL), and unknown alterations.
Distribution of kinase subtypes in Ph-like ALL within each age group.18,19,30,31 Combined prevalence of Ph-like ALL subtypes in childhood NCI standard risk (SR; age 1-9.99 years and white blood cells <50 000/μL), NCI high risk (HR; age 10-15 years or white blood cells ≥50 000/μL), AYAs (16-39 years), and adults (older than or equal to 40 years). Genomic subtypes include IGH-CRLF2, P2RY8-CRLF2, ABL-class fusions (ABL1, ABL2, CSF1R, LYN, PDGFRA, and PDGFRB), JAK2 and EPOR rearrangements, other mutations in JAK-STAT signaling (JAK1/3, IL7R, SH2B3, TYK2, and IL2RB), other kinase alterations (FLT3, FGFR1, and NTRK3,), Ras mutations (KRAS, NRAS, NF1, PTPN11, BRAF, and CBL), and unknown alterations.
Similar to BCR-ABL1 ALL, the incidence of Ph-like ALL increases with age, comprising 10% to 15% of children and over 20% of adults and peaking at 25% to 30% in AYAs.18,19,30 Together with BCR-ABL1 ALL, kinase-driven alterations account for almost one-half of adults with ALL. In children with NCI high-risk ALL and adults, Ph-like ALL is associated with elevated minimal residual disease (MRD) levels and higher rates of treatment failure compared with non–Ph-like ALL patients, with 5-year overall survival rates of 73%, 66%, and 26% in children, adolescents, and adults, respectively.18,19,32 Thus, the high prevalence of Ph-like ALL in AYAs and adults may partly explain the adverse treatment outcomes in these patients. Preclinical studies show that the majority of alterations can be targeted effectively using a combinatorial approach of chemotherapy with ABL1 (eg, dasatinib) or JAK inhibition (eg, ruxolitinib),33 and a number of case studies also report the activity of TKIs used in the treatment of Ph-like ALL patients with refractory disease.32 This approach is currently being tested in frontline studies of patients treated on St. Jude Children’s Research Hospital (Total XVII, NCT03117751) and Children’s Oncology Group protocols (AALL1131, NCT01406756; AALL1521, NCT02723994).32
ETV6-RUNX1 like
Analogous to BCR-ABL1 and Ph-like ALL, ETV6-RUNX1–like ALL is defined by having a gene expression profile and immunophenotype (CD27 positive, CD44 low to negative) similar to ETV6-RUNX1 ALL but lacking the ETV6-RUNX1 fusion.34,35 This subtype is associated with different alterations (gene fusions or copy number alterations) in ETV6, IKZF1, or TCF3, suggesting that global deregulation of lymphoid development is a hallmark of this transcriptional signature. Not surprisingly, ETV6-RUNX1–like ALL is almost exclusively identified in children (∼3%). The prognostic significance is unclear.
DUX4-rearranged ALL
Several studies have identified a rather interesting subtype of B-ALL with a distinct gene expression profile and immunophenotype (CD2 positive) characterized by deregulation of the transcription factors DUX4 and ETS transcription factor (ERG).34,36,37 DUX4 is located in microsatellite D4Z4 repeat domains in the subtelomeric region of chromosome 4, and it is present in 11 to 100 copies on each allele in a normal genome. It is normally expressed in the nucleus of germinal tissues, where its role in gonadal development is unclear.38 In this subtype, translocation of DUX4 to IGH is an early initiating event that results in high expression of a 3′ truncated isoform of DUX4 not normally expressed in B cells. The aberrantly expressed DUX4 binds to an intragenic region of ERG, resulting in expression of an altered ERG transcript, ERGalt, and a truncated C-terminal ERG protein that retains the DNA binding and transactivating domain of ERG, inhibits the wild-type protein, and is transforming in mouse models of B-ALL.37 DUX4-rearranged ALL accounts for 5% to 10% of B-ALL, with a slight peak in AYAs. Of clinical relevance, this subtype is associated with an excellent prognosis in both children and adults, even despite the presence of secondary genetic alterations otherwise associated with poor outcome, such as IKZF1 deletions, which are present in ∼40% of DUX4-rearranged ALL.37
New transcription factors: MEF2D and ZNF384
Multiple 3′ partners have been identified for MEF2D, including BCL9, CSF1R, DAZAP1, FOXJ2, HNRNPUL1, and SS18. All fusions preserve the MEF2D MADS box domain that mediates DNA binding, resulting in enhanced transcriptional activity of MEF2D and a distinct gene expression profile characterized by deregulation of MEF2D targets.39 The exception is MEF2D-CFS1R, which displays the Ph-like gene expression profile.19 Clinically, MEF2D-rearranged ALL is associated with an aberrant immunophenotype (CD10 negative, CD38 positive) and an intermediate to poor outcome.39,40 Deregulation of MEF2D results in the overexpression of histone deacetylase 9, which can be targeted therapeutically using histone deacetylase inhibitors.39
To date, 9 different 5′ fusion partners, usually involving a transcriptional regulator or chromatin modifier, have been identified for ZNF384: ARIDIB, BMP2K, CREBBP, EP300, EWSR1, SMARCA2, SYNRG, TAF15, and TCF3.36,41,42 The entire coding region of ZNF384 is included. Cases with ZNF384-rearranged ALL also have a unique transcriptional signature and are often diagnosed as B-ALL with aberrant expression of the myeloid markers CD13 and/or CD33. Interestingly, a significant proportion of B/myeloid mixed phenotype acute leukemia also harbors ZNF384 rearrangements, suggesting transformation of an early hematopoietic progenitor with multilineage potential.43 An intermediate prognosis has been described in small pediatric cohorts, although larger studies are required to confirm the prognostic relevance.41,42
Recurrent rearrangements of MEF2D and ZNF384 account for ∼4% and ∼5% of children and up to 7% and 10% of AYA patients, respectively. Accordingly, both subtypes are associated with older age of onset (median ages of 14 and 15 years, respectively).39-41
Redefining “other” B-ALL
Despite the advances made in refining the classification of B-ALL, almost one-quarter of cases across the age spectrum lack a subtype-defining lesion and are collectively known as “other.” These cases evade current risk stratification, commonly relapse, and lack targeted therapeutic approaches. To systematically define the spectrum, frequency, and prognostic significance of subtypes across the age spectrum, we recently performed an integrated genomic analysis of almost 2000 B-ALL cases using whole-transcriptome sequencing on all cases and whole-exome or genome sequencing on a subset. In addition to known subtypes, we identified 4 subtypes with distinct gene expression signatures that account for an additional 15% of B-ALL (Zhaohui Gu, Michelle L. Churchman, Kathryn G. Roberts, Ian Moore, Xin Zhou, Joy Nakitandwe, Kohei Hagiwara, Stephane Pelletier, Sebastien Gingras, Hartmut Berns, Debbie Payne-Turner, Ashley Hill, Ilaria Iacobucci, Lei Shi, Stanley Pounds, Cheng Cheng, Deqing Pei, Chunxu Qu, Meenakshi Devidas, Yunfeng Dai, Shalini C. Reshmi, Julie Gastier-Foster, Elizabeth A. Raetz, Michael J. Borowitz, Brent L. Wood, William L. Carroll, Patrick A. Zweidler-McKay, Karen R. Rabin, Leonard A. Mattano, Kelly W. Maloney, Alessandro Rambaldi, Orietta Spinelli, Jerald P. Radich, Mark D. Minden, Jacob M. Rowe, Selina Luger, Mark R. Litzow, Martin S. Tallman, Janis Racevskis, Yanming Zhang, Ravi Bhatia, Jessica Kohlschmidt, Krzysztof Mrózek, Clara D. Bloomfield, Wendy Stock, Steven Kornblau, Hagop M. Kantarjian, Marina Konopleva, Williams Evans, Sima Jeha, Ching-Hon Pui, Jun Yang, Elisabeth Paietta, James Downing, Mary V. Relling, Jinghui Zhang, Mignon L. Loh, Stephen P. Hunger, Charles G. Mullighan, unpublished data, 2018).
IGH rearrangements
The prevalence of rearrangements involving the IGH locus to a range of partners, including CRLF2, CEBP family members (CCAAT/enhancer binding protein), and ID4, is particularly high in AYA and adult ALL (∼10%) and generally confers a poor prognosis.43 In addition to these partners, we identified a subset of cases with pre-B immunophenotype and a unique transcriptional signature characterized by rearrangement of IGH to BCL2, MYC, and/or BCL6 (BCL2/MYC). This subtype is predominantly identified in adults (median age of 48.5 years) and associated with extremely unfavorable outcome. The rearrangements resemble those observed in “double-hit” lymphoma and are rarely identified in ALL.7,44
Nuclear protein in testis midline carcinoma family 1 rearrangements
An additional subtype present exclusively in 1% of childhood ALL (median age of 3 years) involves fusion of almost all of the coding region of nuclear protein in testis midline carcinoma family 1 (NUTM1) to 6 different 5′ partners: ACIN1, BRD9, CUX1, IKZF1, SLC12A6, and ZNF618. NUTM1 is an unstructured nuclear protein exclusively expressed in the testis, and it may play a role in chromatin compaction in developing sperm. Fusions of NUTM1 (commonly BRD4-NUTM1) are a hallmark of nuclear protein in testis midline carcinoma (NMC), an aggressive and fatal subtype of squamous cell carcinoma that also arises frequently in children.45 BRD4-NUTM1 acts to repress differentiation in NMC by recruiting histone acetyltransferases and other transcriptional cofactors to regions of chromatin that are actively transcribing proproliferative and antidifferentiation genes, including MYC.45 Thus, fusions, such as BRD9-NUTM1 in ALL, may have a similar mechanism of action, although experimental studies are required to elucidate the role of NUTM1 fusions in leukemogenesis. In contrast to NMC, ALL patients with NUTM1 rearrangements have an excellent prognosis. Given the involvement of BRD9, it would be interesting to determine the sensitivity of these cells to bromodomain inhibitors.
PAX5-driven subtypes
PAX5 is largely considered to function as a haploinsufficient tumor suppressor in ALL, with secondary heterozygous deletions and loss-of-function mutations present in one-third of all B-ALLs.8 In mouse models, Pax5 heterozygosity cooperates with constitutive activation of the JAK-STAT pathway to promote B-ALL development, supporting its role as a tumor suppressor.46 PAX5 translocations are reported in 2% to 3% of B-ALL.47,48 Our recent analyses identified 2 PAX5 subtypes defined by distinct gene expression profiles and genetic alterations (Zhaohui Gu, Michelle L. Churchman, Kathryn G. Roberts, Ian Moore, Xin Zhou, Joy Nakitandwe, Kohei Hagiwara, Stephane Pelletier, Sebastien Gingras, Hartmut Berns, Debbie Payne-Turner, Ashley Hill, Ilaria Iacobucci, Lei Shi, Stanley Pounds, Cheng Cheng, Deqing Pei, Chunxu Qu, Meenakshi Devidas, Yunfeng Dai, Shalini C. Reshmi, Julie Gastier-Foster, Elizabeth A. Raetz, Michael J. Borowitz, Brent L. Wood, William L. Carroll, Patrick A. Zweidler-McKay, Karen R. Rabin, Leonard A. Mattano, Kelly W. Maloney, Alessandro Rambaldi, Orietta Spinelli, Jerald P. Radich, Mark D. Minden, Jacob M. Rowe, Selina Luger, Mark R. Litzow, Martin S. Tallman, Janis Racevskis, Yanming Zhang, Ravi Bhatia, Jessica Kohlschmidt, Krzysztof Mrózek, Clara D. Bloomfield, Wendy Stock, Steven Kornblau, Hagop M. Kantarjian, Marina Konopleva, Williams Evans, Sima Jeha, Ching-Hon Pui, Jun Yang, Elisabeth Paietta, James Downing, Mary V. Relling, Jinghui Zhang, Mignon L. Loh, Stephen P. Hunger, Charles G. Mullighan, unpublished data, 2018). The first subtype, referred to as PAX5 altered (PAX5alt), comprises cases with diverse PAX5 rearrangements (most commonly to ETV6 or NOL4L), sequence mutations, or intragenic amplification, with the highest prevalence observed in children and AYA (10% each vs 7% in adults) (Figure 3). The second group is defined solely by the PAX5 P80R mutation, which was homozygous in a majority of cases due to deletion of the wild-type PAX5 allele, suggesting that loss of both PAX5 alleles drives the unique gene expression profile in this subtype. The prevalence of PAX5 P80R increases with age, accounting for almost 5% of adults. The prognostic significance of this subtype is still unclear. A high frequency of signaling mutations is identified in patients with PAX5 P80R, particularly in the Ras, JAK-STAT, and other kinase signaling pathways (FLT3 and PIK3CA), highlighting the potential for targeted therapies. Notably, heterozygous Pax5P80R/+ or homozygous Pax5P80R/P80R knock-in mice develop B-progenitor ALL that is transplantable. Interestingly, tumors that arise in Pax5P80R/+ mice genetically inactivate the wild-type Pax5 allele by deletion or truncation, recapitulating the loss of wild-type PAX5 observed in human ALL. In a mouse model of B-ALL, Pax5-Etv6 activated distinct transcriptional pathways, including pre–B-cell receptor signaling and migration/adhesion, confirming its role as an oncoprotein rather than simply acting as a competitive inhibitor of the wild-type Pax5 protein.49 The identification of these PAX5 subtypes as distinct entities highlights the importance of this gene in regulating B-cell differentiation and confirms PAX5 alterations as central initiating events in B-lymphoid leukemogenesis.
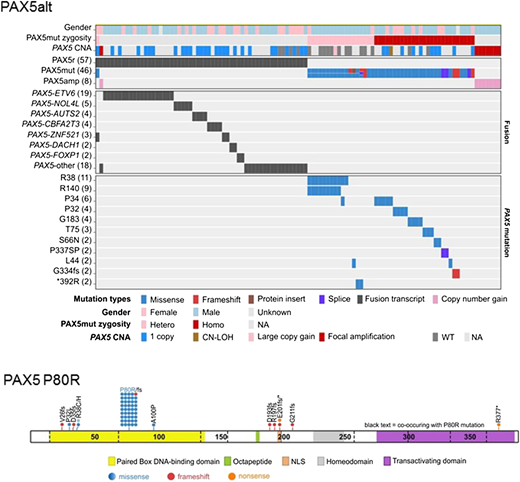
Alterations identified in PAX5alt and PAX5 P80R subtypes. PAX5alt: genetic alterations including gene rearrangements (PAX5r), sequence mutations (PAX5mut), and focal intragenic amplifications (PAX5amp) are shown in the heat map. PAX5 P80R: protein domain plot of PAX5 showing the 57 mutations detected in 44 patients in the PAX5 P80R subtype. CNA, copy number alteration; CN-LOH, copy-neutral loss of heterozygosity; hetero, heterozygous mutation; homo, homozygous mutation; NA, not available; NLS, nuclear localization signal; WT, wild type.
Alterations identified in PAX5alt and PAX5 P80R subtypes. PAX5alt: genetic alterations including gene rearrangements (PAX5r), sequence mutations (PAX5mut), and focal intragenic amplifications (PAX5amp) are shown in the heat map. PAX5 P80R: protein domain plot of PAX5 showing the 57 mutations detected in 44 patients in the PAX5 P80R subtype. CNA, copy number alteration; CN-LOH, copy-neutral loss of heterozygosity; hetero, heterozygous mutation; homo, homozygous mutation; NA, not available; NLS, nuclear localization signal; WT, wild type.
Relapsed ALL
Relapsed ALL responds poorly to conventional therapy and is more common with increasing age. Approximately 20% of childhood ALL patients will experience relapse.2 Although the remission rates for newly diagnosed adult patients are >80% with standard induction regimens, 30% to 60% of these patients will relapse, which carries a very poor prognosis.3 The main curative approach for adults is an allogenic stem cell transplant; however, survival rates for relapsed ALL are improving with the implementation of new immunotherapeutic approaches, including blinatumomab (CD19/CD3 bispecific T-cell engager), inotuzumab ozagamicin (anti-CD22 antibody conjugated to calicheamicin), and CAR T cells (chimeric antigen receptor).50-54
Genomic studies in childhood ALL show that leukemic evolution during relapse usually follows a complex branched pathway, with the majority of primary chromosome translocations retained with the addition of new secondary genetic alterations or enrichment of lesions present in a minor clone at diagnosis.55 Mutations in genes encoding epigenetic regulators and chromatin modifiers are recurrent events in relapsed ALL and can directly influence response to treatment. In particular, mutations in the transcriptional coactivator and acetyl transferase CREBBP occur in up to 20% of relapsed ALL and impair sensitivity to glucocorticoid therapy.56 Recent evidence shows that mutations in 5′-nucleotidase catalytic enzyme II (NT5C2) confer increased resistance to purine analogs at the cost of impaired leukemia cell growth and leukemia-initiating cell activity.57,58 Other recurrent somatic alterations in relapse ALL include deletions of the glucocorticoid receptor NR3C1 and mutations in the H3K36 trimethyltransferase SETD2, the lysine-specific demethylase KDM6A, and the epigenetic regulator MLL2.59
Clearly, certain cytogenetic subtypes within AYA and adult ALL have a dismal prognosis (eg, Ph-like ALL, hypodiploidy, and IGH rearranged); however, the spectrum of secondary lesions acquired during relapse is not well defined. This is of particular interest in the era of immunotherapy, where molecular determinants to response are unknown, and it requires the study of large, uniformly treated cohorts. Enhancing our knowledge of relapse-enriched or acquired alterations is important for initial risk stratification and has implications for molecular monitoring given the increasingly widespread application of deep sequencing approaches to identify low levels of MRD.
Integrating genomic information into the clinic
The potential utility of newly identified genetic alterations in risk stratification and therapy is a key question in the management of patients with ALL. Current risk stratification and treatment algorithms incorporate age, sex, presentation white blood cell count, established cytogenetic alterations, and response to initial therapy measured by levels of MRD. Modulation of treatment intensity based on MRD levels has been an essential factor in improving the outcomes of childhood ALL,60 and it has been incorporated by the National Comprehensive Cancer Network as a recommendation for risk stratification in adult ALL. Because MRD is such a central component of risk stratification, new genomic information should be integrated with response to therapy to develop a comprehensive relapse prediction model.
New genomic data can be incorporated into the clinical management of patients with ALL at several levels. First, sequencing will be increasingly used as a comprehensive molecular diagnostic tool that replaces many currently used diagnostic approaches, such as cytogenetics and fluorescence in situ hybridization. Second, sequencing will identify subtypes with clear prognostic importance not identified by current approaches. This is particularly important in AYAs and adults who lack many of the subtype-defining lesions frequently observed in childhood ALL. Transcriptional profiling is a powerful approach for identifying known and new subtypes; however, its utility as a diagnostic tool may be limited due to complexity of analysis and reproducibility across institutions. Therefore, identifying the driving genomic lesion in each subtype is essential for accurate diagnosis and risk stratification in the clinical setting (eg, DUX4 rearranged and Ph-like ALL). Third, sequencing will identify therapeutic targets or pathways that guide implementation of novel treatment strategies either in frontline studies or after a suboptimal response to initial therapy or relapse. This is exemplified by the identification of Ph-like ALL and the implementation of TKI therapy to improve outcome. Fourth, genomics is likely to have a central role in monitoring responses to therapy by facilitating the early identification of low-level clones that are associated with treatment failure, thereby prompting alternative therapeutic approaches.
Conclusions
Within the last decade, integrated genomic analyses of large cohorts of childhood ALL and more recently, AYA and adult ALL have revolutionized our understanding of the genetic basis of ALL by identifying new subtypes, dysregulated pathways, and therapeutic targets that have led to improved risk stratification and treatment strategies. Despite these advances, a proportion of ALL cases cannot be categorized into any of the currently established subtypes. Ongoing discovery studies are required to fully define the genomic landscape and identify the full repertoire of alterations that contribute to treatment failure and disease relapse.
Correspondence
Kathryn G. Roberts, Department of Pathology, St. Jude Children’s Research Hospital, 262 Danny Thomas Place, MS342, Memphis, TN 38105; e-mail: kathryn.roberts@stjude.org.
