
Abstract
Hydroxyurea has proven efficacy in numerous clinical trials as a disease-modifying treatment for patients with sickle cell anemia (SCA) but is currently under-used in clinical practice. To improve the effectiveness of hydroxyurea therapy, efforts should be directed toward broadening the clinical treatment indications, optimizing the daily dosage, and emphasizing the benefits of early and extended treatment. Here, various issues related to hydroxyurea treatment are discussed, focusing on both published evidence and clinical experience. Specific guidance is provided regarding important but potentially unfamiliar aspects of hydroxyurea treatment for SCA, such as escalating to maximum tolerated dose, treating in the setting of cerebrovascular disease, switching from chronic transfusions to hydroxyurea, and using serial phlebotomy to alleviate iron overload. Future research directions to optimize hydroxyurea therapy are also discussed, including personalized dosing based on pharmacokinetic modeling, prediction of fetal hemoglobin responses based on pharmacogenomics, and the risks and benefits of hydroxyurea for non-SCA genotypes and during pregnancy/lactation. Another critical initiative is the introduction of hydroxyurea safely and effectively into global regions that have a high disease burden of SCA but limited resources, such as sub-Saharan Africa, the Caribbean, and India. Final considerations emphasize the long-term goal of optimizing hydroxyurea therapy, which is to help treatment become accepted as standard of care for all patients with SCA.
Learning Objectives
To understand the rationale and processes involved in establishing optimal hydroxyurea dosing, monitoring, and treatment programs for children and adults with sickle cell anemia
To identify new directions with hydroxyurea therapy, such as personalized dosing, genetic determinants of response, and increased global access
Over the past decade, the decision to recommend hydroxyurea treatment for patients with sickle cell anemia (SCA) has become progressively easier as evidence accumulates regarding the striking laboratory and clinical benefits of early and extended therapy. Since the initial proof-of-principle results were first reported >30 years ago,1 numerous prospective studies have consistently documented the safety and efficacy of hydroxyurea treatment in SCA, covering the full spectrum of ages ranging from adults2-4 to teens and school-age children5-8 to toddlers and even infants.9-11
This report is designed to assist clinicians with optimizing hydroxyurea therapy for their patients with SCA. The goal is to neither provide an encyclopedic review of established or potential benefits and harms nor address treatment barriers related to theoretical long-term risks of carcinogenicity, teratogenicity, and infertility. Clinical experience now documents the safety and efficacy of hydroxyurea for SCA beyond 15 to 20 years of treatment; the remarkably consistent findings of improved laboratory parameters, reduced acute clinical complications, better quality of life, and decreased mortality together form a compelling story that should be told to all patients and families. Accordingly, the goal is to provide new information and concepts regarding the use of hydroxyurea in an attempt to increase its use and effectiveness for patients of all ages. Hydroxyurea therapy should become the standard of care for SCA, and optimizing its use will improve and extend the lives of our patients.
Clinical indications
The National Heart Lung and Blood Institute (NHLBI) of the National Institutes of Health recently issued evidence-based guidelines for sickle cell disease that includes an entire chapter devoted to hydroxyurea therapy.12 Two subsequent publications from the same expert panel include a summary of the main treatment recommendations13 and important knowledge gaps that warrant additional investigation.14 The first treatment recommendation, based on consensus panel expertise, is to educate all patients and families about hydroxyurea therapy. Then moving well beyond the current restricted Food and Drug Administration-label treatment indication, which recommends treatment only for adults with SCA severely affected with at least 3 painful crises in the previous 12 months, the panel provides strong recommendations to treat adults with additional common clinical indications: sickle-related pain that interferes with daily activities or quality of life, severe or recurrent acute chest syndrome, and severe symptomatic chronic anemia. For children, the recommendation is to offer hydroxyurea after age 9 months, regardless of clinical symptoms, and to use shared decision-making in discussions with families.12,15 A final strong and practical recommendation is to use an established prescribing and monitoring protocol in an attempt to maximize the safety and benefits of hydroxyurea therapy.12,13
These treatment recommendations now set a high standard of care for clinicians and patients. No longer is it appropriate or ethical to withhold hydroxyurea discussions until patients are extremely sick or have irreversible disease-related complications. Hydroxyurea therapy should be offered early and routinely as a preventive treatment for SCA, which is a posture entirely consistent with our current clinical approach to other serious hematological disorders, such as hemophilia, thrombophilia, and thalassemia.
Optimal dosing
Although hydroxyurea has proven laboratory and clinical efficacy, the optimal dose to use for individual patients is still a source of considerable debate. The phase 1/2 trial in adults with SCA featured dose escalation and demonstrated a near linear dose response to hydroxyurea, with the treatment dose (mg/kg/d) correlating positively with both the plasma drug concentration and the percentage fetal hemoglobin (%HbF) response.3 These early observations demonstrated the concept and feasibility of stepwise dose escalation toward the maximum tolerated dose (MTD) and further documented that a higher hydroxyurea daily dose provides a greater treatment response.
Most subsequent trials performed in the United States have followed this original approach of hydroxyurea dose escalation to MTD, which can be defined operationally as a stable once-daily dose that leads to the greatest benefits without causing side-effects or toxicities. To achieve this goal, the hydroxyurea dose does not need to be titrated to a particular %HbF threshold or clinical response; instead, the dose can be escalated simply to reach an acceptable nontoxic degree of marrow suppression with target counts for both neutrophils and reticulocytes (Figure 1). Clinical experience across multiple age ranges has shown that the greatest laboratory and clinical benefits occur when hydroxyurea is used at the MTD, which typically averages ∼25 mg/kg/d (Table 1).6-8,10,16-18 However, some patients develop hematological toxicities and do not tolerate 20 mg/kg/d, whereas others can safely take 30 to 35 mg/kg/d. In contrast, several clinical trials from Europe have not featured dose escalation to MTD but instead have used a “clinically effective dose” of 15 to 20 mg/kg/d with good reported clinical outcomes.5,19

Treatment parameters and hematological effects of hydroxyurea therapy at the MTD in 7 cohorts of children with SCA
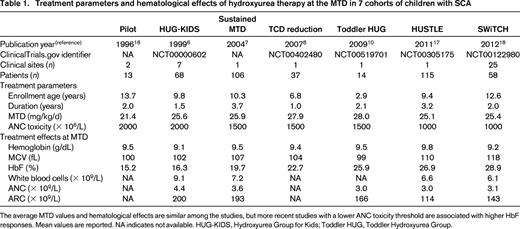
The average MTD values and hematological effects are similar among the studies, but more recent studies with a lower ANC toxicity threshold are associated with higher HbF responses. Mean values are reported. NA indicates not available. HUG-KIDS, Hydroxyurea Group for Kids; Toddler HUG, Toddler Hydroxyurea Group.
To date, there has been no direct comparison of hydroxyurea dosing regimens in SCA to address the question of whether treatment at the MTD provides additional laboratory or clinical benefits or justifies additional risks of toxicity compared with a lower dose. However, when a large number of published clinical trials were analyzed, hydroxyurea treatment at lower clinically effective doses led to an average hemoglobin concentration of <9.0 g/dL, mean corpuscular volume (MCV) <100 fL, and HbF <20%; in contrast, treatment at the MTD yielded better laboratory values with a hemoglobin concentration of >9.0 g/dL, MCV >100 fL, and HbF >20%.20 Because clinical experience has shown that severe hematological toxicity from hydroxyurea is extremely rare, the advantages of MTD appear warranted to yield an optimal treatment response.
Another consideration for optimizing hydroxyurea therapy in SCA is the proper choice of hematological toxicity thresholds to adopt during initial treatment and dose escalation. The absolute neutrophil count (ANC) threshold is the easiest but most critical to define, because transient and reversible neutropenia is the most frequent treatment-related toxicity. In early clinical trials (Table 1), the ANC threshold for temporary treatment discontinuation was conservatively set at 3.0 × 109/L (ANC of 3000/μL) but with clinical experience was subsequently lowered to 2.0 and 1.5, and now the typical ANC threshold to temporarily withhold treatment is 1. 0 × 109/L (ANC of 1000/μL). Table 1 illustrates that studies with lower ANC thresholds led to slightly higher MTD and %HbF values, probably as a result of patients receiving therapy with fewer dosing interruptions. However, a proper MTD also features mild suppression of the reticulocytes, and an absolute reticulocyte count (ARC) of 100 to 200 × 109/L appears to be a safe and reasonable threshold.
A final consideration relates to the optimal dosing of hydroxyurea in very young children, who have lower peripheral blood counts and elevated HbF levels (often >20%) at treatment initiation. As shown in Table 2, the HUSOFT (Hydroxyurea Safety and Organ Toxicity) pilot study used an initial fixed dose of 20 mg/kg/d for infants aged 6 to 24 months,9 which was increased to the MTD in the HUSOFT extension study.21 A similar approach was taken in the phase 3 BABY HUG trial,11 which featured an initial fixed dose of 20 mg/kg/d that was increased in the follow-up studies at the discretion of the individual investigators. Overall, these very young patients have responded very well to hydroxyurea, with excellent treatment responses (hemoglobin concentration of ∼9.0 g/dL, HbF of ∼20%) sustained in these children as they age (Table 2).
Treatment parameters and hematological effects of hydroxyurea therapy for very young children with SCA

In both the HUSOFT and BABY HUG trial, the hydroxyurea dose was 20 mg/kg/d for the main study, but patients were allowed to escalate to the MTD in the extension and follow-up studies. Mean values are reported. NA indicates not available. BABY HUG, Infant Hydroxyurea Group.
Cerebrovascular disease
The onset of cerebrovascular disease in SCA often begins early in life. Even without clinical signs or symptoms, many children and even infants can develop changes within the brain parenchyma and intracranial vessels.8,22 Silent cerebral infarcts23 and overt clinical strokes24 occur most commonly in the first decade of life, and the new NHLBI guidelines recommend that transcranial Doppler (TCD) screening for primary stroke prevention should begin at age 2 years.12
When severe cerebrovascular disease is identified, current evidence supports the use of chronic blood transfusions to prevent progressive disease and especially clinical stroke. Transfusions have proven efficacy for primary stroke prevention in children with either abnormal TCD velocities (>200 cm/s time-averaged mean velocity)25 or silent cerebral infarcts26 and have known benefits for secondary stroke prevention in children with previous stroke. However, indefinite blood transfusion therapy in these clinical settings can lead to serious clinical complications, such as infection, iron overload, transfusion reactions, and erythrocyte alloantibody and autoantibody formation.27 For these reasons, hydroxyurea represents an attractive alternative treatment option to chronic transfusions in the setting of cerebrovascular disease.
Single institution data showed that hydroxyurea treatment lowers secondary stroke rates in children with previous stroke to a rate similar to chronic transfusions: 5.7 recurrent events per 100 patient-years versus 2.2 to 6.4 events per 100 patient-years for published transfused cohorts.28 Based on these encouraging results, the multicenter phase 3 SWiTCH (Stroke With Transfusions Changing to Hydroxyurea; ClinicalTrials.gov identifier NCT00122980) randomized clinical trial compared standard treatment (blood transfusions and iron chelation) to alternate treatment (hydroxyurea and phlebotomy) for secondary stroke prevention and management of iron overload. SWiTCH was stopped early for an equivalent iron concentration in both treatment arms that no longer justified the marginal increased stroke risk; at the time of study closure, 10% of children on hydroxyurea treatment had a recurrent stroke (5.6 events per 100 patient-years), whereas no recurrent strokes occurred on the transfusion arm.18 Risk factors for recurrent stroke included younger age at first stroke, extensive cerebral vasculopathy, and previous transient ischemic attack. Based on the SWiTCH results, chronic transfusion therapy remains the better treatment for children with SCA and previous stroke.
In the current era of TCD screening, the most common indication to commence chronic transfusion therapy in children with SCA is abnormal TCD velocities. Transfusions are efficacious for prevention of primary stroke in this setting, but perhaps hydroxyurea can be used as an alternative treatment option to the indefinite use of blood. Following SWiTCH, the subsequent multicenter phase 3 TWiTCH (TCD With Transfusions Changing to Hydroxyurea; ClinicalTrials.gov identifier NCT01425307) randomized clinical trial compared standard treatment (blood transfusions) with alternate treatment (hydroxyurea) for preservation of TCD velocities and prevention of primary stroke. TWiTCH was recently stopped early for reaching its primary endpoint; data analysis is ongoing, with the main study results due by the end of 2015.
The utility of hydroxyurea for conditional TCD velocities is also under investigation. Based on data that demonstrated that hydroxyurea can lower TCD velocities,8 the SCATE (Sparing Conversion to Abnormal TCD Elevation; ClinicalTrials.gov identifier NCT01531387) randomized clinical trial compared hydroxyurea treatment with standard observation for children with TCD velocities between 170 and 199 cm/s. Unfortunately, SCATE was closed early for slow enrollment, but the truncated study results may provide useful information about the benefits of hydroxyurea in this setting.42 The new EXTEND (EXpanding Treatment for Existing Neurological Disease: ClinicalTrials.gov identifier NCT02556099) trial is currently investigating the benefits of open-label hydroxyurea for children with either conditional or abnormal TCD velocities living in Jamaica, where chronic transfusion therapy is not feasible.
Together, the available published evidence supports the continued investigation of hydroxyurea in the setting of cerebrovascular disease. Although blood transfusions may be superior in most settings, challenging clinical scenarios are frequently encountered (eg, multiple erythrocyte alloantibodies, severe iron overload, or religious objection to blood transfusions), in which hydroxyurea could be an effective and even preferred treatment option for cerebrovascular disease in children with SCA. Especially in limited-resource countries without a safe and adequate blood supply, hydroxyurea may represent a clinically useful and cost-effective therapeutic strategy for preventing stroke.29
Switching from transfusions to hydroxyurea
For patients with SCA who receive chronic transfusions, an important therapeutic goal is to maintain the total hemoglobin concentration high enough to suppress endogenous sickle erythropoiesis; operationally, this means maintaining ≤30% HbS, which is typically measured just before each transfusion. Although this 30% HbS target has never been tested formally in a randomized clinical trial, it has both a physiological basis and many years of clinical effectiveness. In actual practice for stroke prevention, most academic practices can achieve values close to this goal; trough HbS levels in children transfused either for primary or secondary stroke prevention average ∼35%.30,31 Because transfusions are clinically effective, the decision to pursue a “therapeutic switch” from a familiar transfusion regimen to a relatively unfamiliar hydroxyurea protocol must be considered carefully, especially when the patient has an increased risk of stroke.
In designing a switch from transfusions to hydroxyurea, three key principles should be recognized: (1) an overlap period of dual therapy is desirable, because the benefits of transfusions are immediate, whereas those of hydroxyurea have a slower onset; (2) transfusions should be weaned gradually to allow endogenous erythropoiesis to occur under the influence of hydroxyurea treatment; and (3) hydroxyurea should reach a stable MTD with documented hematological benefits before transfusions are discontinued. To accomplish this switch efficiently and safely, we have developed a procedure that simultaneously weans transfusions while escalating hydroxyurea to MTD, based on experience with >150 children who have undergone this therapeutic transition (Figure 2). The most critical feature is weaning the transfusions with stepwise lowering of hemoglobin targets, while increasing the hydroxyurea dose progressively to a stable MTD32 ; in children with SCA who are adherent and achieve a stable MTD, the transition can be accomplished safely in ∼6 to 8 months.18,28

Hydroxyurea and phlebotomy
An important consequence of chronic transfusion therapy is iron overload, which occurs from the accumulation of transfused erythrocytes. Transfusion-acquired hemosiderosis develops as the transfused red blood cells age and undergo phagocytosis within the reticuloendothelial system. Initially, the recycled iron is stored primarily within tissue macrophages but can eventually deposit in the parenchymal cells and cause organ damage. Therefore, a typical chronic transfusion regimen includes periodic monitoring of total body iron stores, coupled with chelation therapy designed to remove excess iron and ameliorate its deleterious effects.
When patients with SCA switch from chronic transfusions to hydroxyurea therapy, the problem of hemosiderosis persists, so the management of transfusional iron overload is an important consideration. However, the optimal means by which to normalize iron burden in this setting has not been established. Effective oral chelators are now available, including deferasirox and deferiprone, but their use with hydroxyurea has not been evaluated carefully. Concomitant use of chelators with hydroxyurea could potentially lead to organ toxicity or side-effects that reduce compliance. In contrast, serial phlebotomy represents a safe and effective alternative approach to chelation therapy for removing the iron burden. Repeated phlebotomy procedures are well-accepted and well-tolerated in the clinical settings of polycythemia vera, hereditary hemochromatosis, posttransplantation, and rare blood antigen disorders. Perhaps the greatest challenge of serial phlebotomy in patients with SCA is the underlying anemia, but hydroxyurea therapy at a stable MTD typically raises the hemoglobin concentration to >9.0 g/dL, which allows the procedure to proceed safely.33
With proper planning and an established process, outpatient phlebotomy can commence soon after transfusions have been discontinued. Several key concepts are critical for successful therapeutic phlebotomy: (1) proper preparation of the patient and training of all medical staff are essential; (2) actual volumes and speed of blood removal are not as important as slow and steady progress over time; (3) adherence with hydroxyurea at MTD is necessary to maintain safe hemoglobin levels; and (4) normalization of iron stores through phlebotomy will take approximately the same amount of time as the previous duration of chronic transfusions. Once phlebotomy commences, the frequency and volumes of blood removal can be individualized, but 5–10 mL/kg/month is well-tolerated by most patients with SCA (Figure 3). In prospective clinical trials, we have guided several thousand phlebotomy procedures in >100 children with SCA.28,33 The vast majority of these procedures were completed safely without any side-effects; with sufficient time, serial phlebotomy can normalize iron stores.28

Future directions
An important and challenging problem related to hydroxyurea dosing in SCA is the time and effort required to achieve a stable MTD. Historically, the stepwise approach of dose escalation requires >6 months, during which time the patient typically receives monthly monitoring while on a suboptimal dose without a full clinical benefit. Because data from the HUSTLE (Hydroxyurea Study of Long-Term Effects; ClinicalTrials.gov identifier NCT00305175) trial demonstrated that patients receiving hydroxyurea have variable pharmacokinetics and pharmacodynamics,17 treatment regimens could be designed with predictive models based on a patient's individual characteristics. The NDEPTH (Novel Dose Escalation to Predict Treatment with Hydroxyurea; ClinicalTrials.gov identifier NCT02042222) clinical trial uses baseline creatinine, ARC, and body mass index, which were among the simplest parameters that best predicted the hydroxyurea MTD.17 In contrast, the newly opened TREAT (Therapeutic Response Evaluation and Adherence Trial; ClinicalTrials.gov identifier NCT02286154) uses first-dose pharmacokinetic parameters to generate the optimal hydroxyurea dose. Together, these two clinical trials should advance the field with a goal of “personalized dosing” and shorten the time to MTD, which will help optimize the hydroxyurea effect.
Another exciting research opportunity is predicting the HbF response to hydroxyurea therapy. Although there is no absolute %HbF threshold required for clinical benefit, the “more is better” axiom is accurate for HbF levels in SCA. All children who are adherent to hydroxyurea treatment will respond with an increased %HbF, but the magnitude of the HbF response is difficult to predict. Candidate genes that influence baseline %HbF are associated with higher HbF responses to hydroxyurea,17,34 and novel genetic variants influencing HbF responses have been proposed recently.35 However, additional validation studies are required before these or other single nucleotide polymorphisms can be proposed as accurate predictors of hydroxyurea treatment response.
Soon after the NHLBI evidence-based guidelines for SCA were promulgated, a subsequent manuscript from the expert panel outlined specific knowledge gaps. One important gap was the lack of data regarding the risks and benefits of hydroxyurea for patients with non-SCA genotypes, especially HbSC disease or HbS/β+ thalassemia.14 Small series and anecdotal evidence suggest that hydroxyurea may have benefits for these less common genotypes, and many clinicians now report “clinical drift” regarding the use of hydroxyurea treatment for these patient populations. However, because the pathophysiology of these genotypes is quite different from HbSS, the potential benefits and harms of hydroxyurea therapy also differ. Accordingly, a definitive phase 3 randomized clinical trial is warranted; especially for patients with HbSC disease, a prospective multicenter clinical trial comparing low-dose hydroxyurea versus MTD would greatly advance the field.
Similarly, the benefits and harms of hydroxyurea treatment for women with SCA during pregnancy and lactation represent an important knowledge gap that is becoming increasingly clinically relevant.14 Women with SCA of child-bearing age are now more likely to receive hydroxyurea therapy, but the current recommendation is to discontinue treatment in the setting of either pregnancy or lactation, which represents an ethical challenge for both the mother and the infant. An argument can be made that effective disease-modifying therapy for a mother with SCA improves her own health and that of her unborn baby and that the known benefits of hydroxyurea outweigh the theoretical risks. However, because the hydroxyurea package insert provides a strongly worded caution against its use in these settings, the NHLBI evidence-based guidelines also include this recommendation to discontinue hydroxyurea during pregnancy and lactation, although the panel noted very low-quality evidence.12 Women with SCA and hydroxyurea exposure have had successful normal pregnancies,36 and no clear teratogenic phenotype exists for hydroxyurea in humans, but more data should be collected. A registry of women with SCA who choose to continue hydroxyurea treatment during pregnancy or lactation should be established to provide accurate prospective data regarding the risks and benefits of continuing hydroxyurea in this important setting. A similar case can be made for men of child-bearing years, with regard to monitoring the effects of hydroxyurea on male fertility, recognizing that a mildly decreased sperm count (20-50 × 106/mL) is still well within the normal range and does not necessarily confer infertility.37
Global access
A tremendous future opportunity for optimizing hydroxyurea therapy lies with its introduction and expansion into the global arena. Most of the current burden of SCA exists within limited-resource countries, especially sub-Saharan Africa and India,38 whose global burden will become even greater by 2050.39 The World Health Organization has issued directives to the African Region for the development of national sickle cell strategies that include sickle cell awareness, early detection, and the establishment of care and treatment programs.40 Hydroxyurea is arguably an ideal disease-modifying therapeutic option for SCA in this setting; however, its safety, feasibility, and benefits have not been demonstrated in most limited-resource countries to date. In India, low-dose hydroxyurea was reported to be safe, practical, and effective therapy for SCA.41
Several important clinical trials are currently investigating the use of hydroxyurea in Africa, including the following: (1) SPIN (Stroke Prevention in Nigeria; ClinicalTrials.gov identifier NCT01801423), a feasibility trial that will assess daily adherence to hydroxyurea; (2) NOHARM (Novel use Of Hydroxyurea in an African Region with Malaria; ClinicalTrials.gov identifier NCT01976416), a randomized placebo-controlled trial investigating the severity of both malaria and SCA; and (3) REACH (Realizing Effectiveness Across Continents with Hydroxyurea; ClinicalTrials.gov identifier NCT01966731), a pilot safety trial of open-label hydroxyurea in four locations within Central and East Africa. With the data collected from these and other trials, hydroxyurea use in limited-resource settings will likely expand so that many more children and adults with SCA can benefit from this therapy.
Final considerations
Optimizing hydroxyurea therapy for patients with SCA will not require herculean efforts or total revamping of our current management practices but will require some pragmatic changes in our collective approach to this disease. First, clinicians should recognize the merits of hydroxyurea as preventive treatment to avoid disease complications and embrace shared decision-making with patients and families; this philosophy is well-accepted in many hematological disorders but is not yet embraced for SCA. Second, the clinical indications for treatment should be broadened to include less severe and even asymptomatic young patients; this relatively simple modification fits well with the newly published NHLBI guidelines and will provide treatment for many more patients. Third, whenever safe and feasible, patients should be managed with established protocols that use dose escalation to MTD with appropriate monitoring, which helps ensure a safe and effective dose. Eventually, the long-term goal of optimizing hydroxyurea therapy is to help treatment become accepted as the standard of care for all patients with SCA. Hydroxyurea is neither a cure nor a panacea, but it does significantly reduce morbidity and mortality in SCA. Until better and more effective treatments are developed or until a safe and affordable cure becomes available, clinicians should use hydroxyurea “early and often” to improve the lives of children and adults with SCA.
Correspondence
Russell E. Ware, Division of Hematology, Department of Pediatrics, 3333 Burnet Ave, Cincinnati Children's Hospital Medical Center, Cincinnati, OH 45229; Phone: 513-803-4597; E-mail: russell.ware@cchmc.org.
