
Abstract
Treatments for patients with SCID by hematopoietic stem cell transplantation (HSCT) have changed this otherwise lethal primary immune deficiency disorder into one with an increasingly good prognosis. SCID has been the paradigm disorder supporting many key advances in the field of HSCT, with first-in-human successes with matched sibling, haploidentical, and matched unrelated donor allogeneic transplantations. Nevertheless, the optimal approaches for HSCT are still being defined, including determining the optimal stem cell sources, the use and types of pretransplantation conditioning, and applications for SCID subtypes associated with radiosensitivity, for patients with active viral infections and for neonates. Alternatively, autologous transplantation after ex vivo gene correction (gene therapy) has been applied successfully to the treatment of adenosine deaminase–deficient SCID and X-linked SCID by vector-mediated gene addition. Gene therapy holds the prospect of avoiding risks of GVHD and would allow each patient to be their own donor. New approaches to gene therapy by gene correction in autologous HSCs using site-specific endonuclease-mediated homology-driven gene repair are under development. With newborn screening becoming more widely adopted to detect SCID patients before they develop complications, the prognosis for SCID is expected to improve further. This chapter reviews recent advances and ongoing controversies in allogeneic and autologous HSCT for SCID.
Learning Objective
To understand current approaches to the treatment of infants with SCID, including issues related to genetic etiology, donor choice, conditioning regimen, and the use of gene therapy
SCID
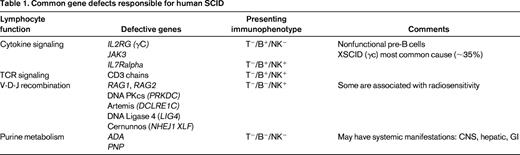
SCID is the most severe primary immune deficiency, with absent T- and B-cell function (and sometimes NK, depending on the responsible affected gene; Table 1). All forms of SCID have in common high early mortality from infections without treatment. Allogeneic BM transplantation can be curative, with ≥90% successful outcomes with HLA-matched sibling donors, but lower survival and variable immune reconstitution with alternative donors (mismatched family member or unrelated adult or cord blood). SCID has been a guiding disorder for the fields of hematopoietic stem cell transplantation (HSCT) and gene therapy as the initial setting for allogeneic human BM transplantation (1968), use of matched unrelated donor BM (1974), and transplantation with haploidentical (parent) BM (1976), and was the first disease approached by gene therapy (1990) and, arguably, the first “cure” by gene therapy (1999).
Role of pretransplantation conditioning in HSCT for SCID
Despite the successes that have been achieved in the treatment of SCID patients, there remain many areas of ongoing controversy concerning optimal approaches. Perhaps most contentious is the use and type of pretransplantation conditioning.1 HSCT products [BM, umbilical cord blood (UCB), peripheral blood stem cells) are heterogeneous, with long-lived stem cells present at low frequencies and lineage-restricted progenitor cells of multiple stages and activity at higher frequencies. Immune reconstitution may result from combined effects of short-term lymphoid progenitor cells producing large numbers of T, B, and NK cells early, and rarer long-term stem cells serving as a sustained source of lymphocytes, especially B and NK cells. Without conditioning, only the former effect may be achieved from T-lymphoid progenitors, supporting T-cell function reconstitution, but with minimal B and NK function reconstitution due to absence of stem cells. In fact, a recent study reported that BM from SCID infants [X-linked SCID (XSCID), Jak3-deficient, and adenosine deaminase (ADA)-deficient] contained essentially normal levels of early lymphoid committed progenitors, and these may be among the donor (or autologous gene-modified) cells that re-engraft to support T-lymphoid reconstitution in these nonconditioned SCID patients.2 With conditioning, both the effects on early T-cell recovery from progenitors and the sustained support of B and NK activity from engrafted multipotent, long-term HSCs may be achieved.
The optimal conditioning regimen, if one is to be used, remains to be determined. Transplantations for SCIDs have been done using full-intensity conditioning [eg, busulfan/cyclophosphamide, busulfan/fludarabine ± antithymocyte globulin (ATG), alemtuzumab, melphalan], reduced-intensity conditioning (eg, fludarabine/ATG, “little Bu,” treosulfan), or just serotherapy (eg, ATG, alemtuzumab). As in other transplantations, reduced intensity often equates to reduced toxicity, but may also be associated with reduced efficacy. Other confounding variables that may affect success include the presence of recipient NK function (which may mediate graft rejection), infants with engraftment of maternal T cells, or with autologous auto-reactive T cells as in Omenn's syndrome, and SCID-related radiosensitivity (Artemis, DNA PKcs defects); in these cases, standard conditioning can have devastating late effects.3 To a large extent, the choice of conditioning has related to the choice of HSC source, with conditioning more likely to be used with UCB, variably with adult unrelated donors, less often with haploidentical donors, and almost never with matched sibling donors. The molecular defect causing SCID may also influence the choice of conditioning regimen, if used, with the presence of active NK cells, as in recombinase-deficiency etiologies (T−/B−/NK+) sometimes taken as an indication for conditioning to overcome allograft resistance.
The well-known toxicities of chemotherapy are potentially heightened when these agents are administered to young developing infants, especially for long-term effects on growth and development, the CNS, endocrine, reproductive, skeletal, dental and other systems. SCID patients presenting with infections may have their condition worsened from the conditioning-induced neutropenia added to the preexisting lymphocyte defects, or from administration of posttransplantation GVHD prophylaxis. Overall survival rates are high for SCID infants treated without conditioning and most do achieve sufficient T-cell function to avoid the lethality of SCID.
However, there is clearly a higher frequency of HSC engraftment from “making space” with pretransplantation conditioning. Nonconditioned HSCT leads to minimal engraftment of HSC, which may cause long-term deficiencies in B cells (antibody production) and NK cells. Improved stem cell engraftment likely leads to better B-cell reconstitution, allowing gamma-globulin replacement to be stopped, and may also support IgA production and better mucosal immunity.1 Because the proponents and opponents of conditioning are convinced of the correctness of their approach, it is unlikely that a definitive randomized trial will be performed to address this critical issue. However, conclusions may be derived from compiled data from multiple centers, as is being done in the newly developed Primary Immune Deficiency Treatment Consortium (PIDTC) (see PIDTC section) and by the European Society for Immunodeficiency (ESID), the Inborn Errors Working Party of the European Group for Blood and Marrow Transplantation (EBMT), the Center for International Blood and Marrow Transplant Research (CIBMTR), and others.
HSC source for SCID patients lacking HLA-matched sibling donors
A recent publication from European centers analyzed the outcomes for SCID patients treated with different graft types.4 The estimated 5-year overall survival rates were not significantly different when using mismatched related donors (MMRD; haploidentical) or UCB: 62% ± 4% after MMRD and 57% ± 6% after UCB. The conclusion of this study was that, for children with SCID and no HLA-identical sibling donor, either UCB or MMRD represent valid HSC sources for transplantation with quite similar outcomes based on treating center expertise.
Graft engineering to reduce GVHD risks
The optimal method for T-cell depletion of mismatched related (haploidentical) donor BM is another area of debate. Some centers perform T-cell depletion by the first clinically acceptable method that was developed, with E-rosetting of T cells with sheep RBCs after removing cells clustered by soybean agglutinin (SBA−/E+). T-cell depletion that involves the use of these biological reagents may have unexpected benefits, by leaving some cell types in the graft that facilitates engraftment of the HSC and by removing donor B cells that may carry EBV, making posttransplantation lymphoproliferative disease rare. Many other centers are using immunoaffinity with monoclonal antibody columns to CD34+ cells to enrich for stem and progenitor cells. This approach does effectively remove T cells, but may also eliminate other useful beneficial cell populations. More selective graft manipulation is being studied, such as depletion of TCR alpha/beta (+) T cells (potential source of GVHD effectors) and CD19+ B cells (potential to transmit EBV), with retention of myeloid, NK, and gamma/delta T cells, as well as HSCs.5 Use of peritransplantation serotherapy, such as ATG or alemtuzumab, may also reduce alloreactivity, but may impede initial immune reconstitution.
PIDTC
An important development for the field has been the establishment of the PIDTC, a collaborative program with membership by most of the major transplantation centers in North America that perform HSCT for SCID and other PID (http://rarediseasesnetwork.epi.usf.edu/PIDTC/).6 Funded as an National Institutes of Health Rare Diseases Clinical Research Network (RDCRN), this collaborative group of investigators have established protocols to ascertain clinical and laboratory data retrospectively on all SCID patients identified in the past 4 decades and to record prospectively a common dataset on all SCID patients treated after 2009 [other protocols will do the same for chronic granulomatous disease (CGD) and Wiskott-Aldrich syndrome (WAS)]. The ESID/EBMT has been performing analyses like these for several decades, producing some of the most informative reports on outcomes for a large series of SCID infants.4,7,8
In addition to publications proposing standards of care for the diagnosis of SCID and its pretransplantation medical management,9 a first major PIDTC study has been completed consisting of a retrospective analyses of outcome for 240 SCID patients diagnosed between 2000 and 2009.10 As seen in many other studies, outcomes were best for recipients of matched sibling donor transplantations. Infants treated at <3.5 months of age did well with all donor types (matched sibling, UCB, matched unrelated, and haploidentical), whether given conditioning or not. Older subjects also did well if they were free of infections at the time of transplantation. In contrast, survival for patients with active infections at transplantation was better in the absence of conditioning. However, immune reconstitution (T-cell numbers, naive T cells, B-cell function) was better for recipients who were conditioned.
Newborn screening for SCID
Several retrospective analyses showed significantly better outcome for SCID infants diagnosed early (eg, 2nd affected child in family), before onset of infections, failure to thrive, and other SCID-related complications that may occur before implementation of protective isolation and other intensive medical care.11-13 Based on these observations, it was hypothesized that newborn screening (NBS) would afford the opportunity to intervene with a transplantation before the complications of SCID have occurred, improving survival and other outcomes. Efforts were made to develop SCID screening tests that would have the necessary high sensitivity and specificity and low cost for population-wide applications. Quantitative PCR measurement of TCR excision circles (TREC) from a small portion of the dried blood spots taken from neonates for screening emerged as most suitable.14 In May 2010, the US Secretary of Health and Human Service Kathleen Sebelius accepted the recommendation of the Secretary's Advisory Committee on Heritable Disorders in Newborns and Children (SACHDNC) to add SCID to the standard panel of conditions screened for in newborns. However, this recommendation is being implemented on a state-by-state basis. Although there has been a progressive increase in the numbers of states screening for SCID, with >50% of US births occurring in a state performing screening by 2014, each state has developed its own methodology to measure TREC, defined the cutoff values to trigger further immunological investigations, and established the follow-up paradigms.
The incidence of SCID measured in California in the first 2 years after the implementation of screening was 1/54 000 from almost 1 million births screened, and 1/58 000 in a compilation of findings from multiple US states performing NBS.15 These frequencies of SCID in the United States are ∼2-fold higher than the previous referral-based estimates of 1/100 000; the discrepancy implies that a significant number of infants born with SCID before the advent of screening did not survive to be diagnosed, leading to underestimation of its frequency. Another finding from the screening results to date is that the distribution of SCID genotypes is different from that reported in some prior large, single-center series,16 with RAG1/2 defects much more common than previously detected (∼1/3 of identified genotypes), making XSCID and ADA-deficient SCID responsible for a relatively smaller percentage of cases (∼35% and 10%–15% respectively).
NBS for SCID is presenting several new opportunities and challenges. The best approach to treating these infants remains to be determined. Because they are being diagnosed within the first few weeks of life, earlier transplantation is possible; however, toxicities from cytoreductive conditioning regimens may be heightened at this early age in terms of adverse effects on growth, neurodevelopment, endocrine function, etc. Therefore, the PIDTC is planning a prospective clinical trial that will evaluate the role of low-dose busulfan cytoreductive conditioning in a gradual dose escalation schema to improve donor cell engraftment and hence the acquisition of B-cell function adequate to obviate the need for life-long gammaglobulin replacement.
A significant number of infants with T lymphopenia of uncertain etiology (not attributable to a known SCID gene defect or some other recognized cause such as Di George syndrome) are being identified as “screen failures” in NBS. The causes and natural histories of these disorders are unknown, as are the roles for transplantation or other interventions. Members of the USIDnet, the Clinical Immunology Society and the Immune Deficiency Foundation, are collaborating to develop a registry for these individuals to follow their clinical courses prospectively.
Autologous HSCT/gene therapy
Gene therapy as conceived for SCID and other genetic blood cell diseases is basically an autologous HSCT with ex vivo gene modification of the graft. Use of autologous HSCs would allow each patient to serve as their own donor and obviate the major immunological barriers seen with allogeneic HSCT, especially graft rejection and GVHD. SCID was identified early as a favorable disease candidate for gene therapy via autologous HSCs based on the success with nonconditioned allogeneic transplantations using matched sibling donor BM, in which even undetectable engraftment of HSCs (based on absence of detectable donor myeloid cells, erythroid cells, or HSCs), results in sustained T-cell, and sometimes B-cell and NK-cell, reconstitution. This implied a “selective advantage” for gene-corrected cells, leading to their amplification from even a low number of gene-corrected stem cells.
The central technical challenge is that gene correction must be performed in a large number (>2 × 106/kg CD34+ cells) and the highest possible percentage of the relatively fragile HSC collected from a patient's BM, UCB, or peripheral blood. HSCs will proliferate to produce millions of mature blood cells; therefore, permanent gene correction of HSCs requires stable persistence of the therapeutic gene with cell division. Current options for stable gene modification include: gene addition using vectors made from the Retroviridae family, including retroviral vectors (γ-RVs; eg, MLV), lentiviral vectors (eg, HIV, SIV, EIAV), or Spumaviral vectors (eg, Human Foamy Virus); or the use of methods for direct in situ gene correction of the defective endogenous gene. Effective methods to expand HSCs after gene correction would be useful.
A major limitation for the use of γ-RVs for gene therapy is that they only are capable of integrating into the genome of dividing cells, but most of the HSCs with long-term cell production activity are quiescent. Therefore, major efforts were made to identify hematopoietic growth factors that will stimulate HSCs to divide without inducing terminal differentiation and loss of stem cell self-renewal activity. A relatively standard mixture of cytokines has been used by most investigators, including ckit ligand, flt-3 ligand, thrombopoietin (and sometimes IL-3, although there is no evidence that it adds or detracts to HSC transduction17 ). Lentiviral vectors are less dependent on stimulation of CD34+ cell populations for effective transduction than are γ-RVs, but the extent of gene transfer is significantly increased when cells are prestimulated in the same cytokine combinations before transduction.18 Typical clinical cell-processing protocols for transduction of CD34+ cells with lentiviral vectors entail 1 day of prestimulation and 1 day of transduction, compared with protocols for γ-RVs with 2 days of prestimulation and 2-3 days of transduction; the shorter culture for lentiviral transduction may better preserve stem cells and enhance engraftment polyclonality.
ADA-deficient SCID
Deficiency of the purine metabolic enzyme ADA was the first genetic cause of human SCID to be identified* and to have the responsible normal gene cloned.19 ADA SCID was thereby the first disorder to be addressed with gene therapy, with trials in the 1990s using γ-RVs carrying human ADA cDNA targeting either peripheral blood T cells or HSCs from BM or UCB. No significant clinical efficacy was seen, with relatively low levels of gene-modified cells in the subjects. Most continued to receive PEG-ADA enzyme replacement therapy that supported immune function but may have obscured potential effects from the transplantation.
During that decade, improved methods for producing retroviral vectors and for the manipulation of HSCs in vitro were developed that were applied to subsequent trials. The major advance in the pioneering studies at San Raffaele-Telethon Institute for Gene Therapy (TIGET) was the adoption of nonmyeloablative condition with busulfan (4 mg/kg) before the transplantation of the ADA gene-modified CD34+ cells.20 The initial report of immune reconstitution in 2 subjects was extended to at least 18 subjects treated, with successful immune reconstitution in most, which has persisted with no late adverse events.21 Similar trials at University College London (UCL)/Great Ormond Street Hospital (GOSH) and University of California, Los Angeles (UCLA)/National Institutes of Health (NIH) have confirmed similar responses using retroviral vectors and similar low-dose busulfan conditioning.22-24
In total, at least 40 ADA-deficient SCID patients have been enrolled in these trials of autologous transplantation of ADA gene-corrected BM CD34+ cells. The majority developed protective immunity and remain well without other interventions, such as enzyme replacement or allogeneic HSCT. In our studies of 20 ADA-deficient SCID subjects, we have observed the most robust levels of immune reconstitution in infants treated at <1 year of age, and less improvement of immune function in older subjects (eg, 4-15 years). In addition to the greater thymic potential in the younger subjects contributing to the better responses, higher numbers of CD34+ cells per kilogram have generally been isolated from the BM of younger subjects and, therefore, their grafts had larger cell doses than for the older subjects. Importantly, there have been no vector-related complications in any of the ADA-SCID patients, in sharp contrast to the multiple leukoproliferative complications seen in several other clinical trials targeting HSCs with γ-RVs for CGD and WAS (see XSCID section).
Investigators at TIGET performed integration site analyses in blood cells across their different trials for ADA SCID that used MLV vectors targeting either peripheral blood T cells or BM HSCs.25 After transduction and transplantation of either transduced T cells or HSCs, there was similar enrichment for integrants within gene-rich regions across the genomes of circulating blood cells years after gene therapy. However, integrants in transduced T cells were enriched for those near genes annotated as being related to immune function and T-cell functions and pathways; that is, genes that would be highly expressed in the targeted T cells. Transduced HSCs did not show the bias toward these lymphoid-related sites. These investigators had previously shown that subjects who received γ-RV-modified CD34+ BM transplantation did have some clustered integrants in the LMO2 and Evi1 loci, but no clonal expansion.26
New trials have begun in the United Kingdom and the United States based on similar clinical trial designs with low-dose busulfan, but using a lentiviral vector (www.ClinicalTrials.gov identifiers #NCT01380990 and #NCT01852071). EFS-ADA produced at UCL/GOSH is a self-inactivating (SIN) lentiviral vector carrying a codon-optimized human ADA cDNA under transcriptional control of the human Elongation Factor 1-alpha gene “short” promoter. Preclinical studies performed jointly by the UK and US investigators showed that the EFS-ADA vector was produced at high titer, efficiently transferred the ADA cDNA to human BM CD34+ cells assayed in vitro and by NSG xenografts, expressed ADA enzyme activity at a level similar to that of the γ-RV used previously, and had a safer profile in the in vitro immortalization assay.17,27 Enrollment began into the trials at UCL/GOSH in 2012 and at UCLA/NIH in 2013.
XSCID
The results of the first clinical trials of gene therapy for XSCID using γ-RVs to carry a normal human IL2Rg (γc) cDNA in Paris, France, and London are well known.28-30 Significant T-cell immune reconstitution occurred in the majority of subjects, only limited in those with severe preexisting infections at the time of transplantation. B-cell functional recovery was less consistent, although half of the subjects have sufficient B-lymphoid activity to be able to discontinue gamma-globulin replacement. The earliest treated subject is out 15 years with sustained normal T-cell counts, continued production of new T cells based on levels of TREC, and with polyclonal T-cell repertoire based on T-cell receptor spectratype analysis.
However, 5 of 20 subjects between the 2 trials developed T-lymphoid leukemia-like leukoproliferative complications 2-5 years after gene therapy.31-33 This proved fatal in 1 case, but was reversible in 4 with ALL-type therapy; there have been no new cases occurring after 5 years. The cause is the process of insertional oncogenesis by which strong enhancer elements in the long-terminal repeats (LTR) of an integrating vector can trans-activate adjacent protooncogenes (eg, LMO2, Evi1), leading to clonal expansion and ultimately malignant transformation. Despite these serious complications, 90% of the subjects are surviving, with 85% having sustained immune reconstitution, results at least equivalent to those using HLA-matched related donors.
Similar leukoproliferative complications have occurred in trials using γ-RVs for HSC gene therapy for CGD and WAS34,35 ; ADA is an exception, with no leukoproliferative complications in >40 treated subjects. The basis for this difference is unknown; speculations include some protection in ADA SCID due to the inherent myeloid abnormalities that have been reported in ADA SCID,36 theoretically raising the transformation threshold, or lower proliferative stress on the gene-corrected lymphoid progenitors due to the metabolic cross-correction effect of ADA-replete cells on ADA-deplete cells.
Multiinstitutional phase 1/2 trials are evaluating the treatment of SCID-X1 patients with a putatively safer SIN γ-RV vector (lacking LTR enhancers) carrying the IL2Rγ cDNA under control of the EFS promoter (www.ClinicalTrials.gov identifier #NCT01410019, NCT01175239, and NCT01129544). Common clinical protocols and vector production lots are being used. As with the earlier trials, the target cell is BM CD34+ cells and no conditioning is being given. The trials are being done under separate regulatory oversight (France, United Kingdom, United States), but the data combined for analyses. Initial reports have described the results with 9 treated subjects.37 Immune reconstitution similar to that seen with earlier trials has been achieved with the SIN γ-RVs, but with significantly fewer clustered vector integrants in genes implicated in leukoproliferation, such as LMO2 or Evi1. No clonal expansions or clinical leukoproliferative events have occurred to date. Again, there has been incomplete or minimal B-cell and NK-cell reconstitution, likely due to the avoidance of conditioning.
Brian Sorrentino et al at St. Jude Children's Research Hospital (SJCRH) have developed a SIN lentiviral vector carrying a normal human IL2Rγ cDNA under control of the EFS promoter for the treatment of XSCID.38 A novel stable packaging cell line was derived at SJCRH and used to produce the clinical-grade vector.39 Extensive preclinical safety studies were done that demonstrated undetectable genotoxocity from the vector, in contrast to the γ-RVs with intact LTR used as a positive control.38 They have opened a clinical trial to use the lentiviral vector to transduce BM CD34+ cells from XSCID infants to be given without conditioning (www.ClinicalTrials.gov identifier #01512888).
Harry Malech et al opened a clinical trial at the NIH Clinical Center that uses the same lentiviral vector from SJCRH, but focuses on a different subject population: older males with XSCID who had prior transplantations but did not achieve satisfactory immune reconstitution (www.ClinicalTrials.gov identifier #01306019). Although a limited cohort, there are several boys and young men who had nonconditioned haploidentical transplantations for XSCID in prior years and have had some T-cell immunity, which has been waning, and minimal B-cell or NK-cell reconstitution. They have been given reduced-intensity conditioning (busulfan 6 mg/kg) followed by autologous peripheral blood stem cells modified with the IL-2Rγ lentiviral vector. Early studies demonstrate modestly increased numbers of T, B, and NK cells, with clinical improvement of protein-losing enteropathy in one patient with increasing IgG levels.
Each genetic etiology of SCID requires a dedicated research attack to develop the appropriate vectors, to demonstrate disease-modifying activity in preclinical studies, and to develop and perform clinical trials. Studies are in progress to develop vectors for the correction of RAG1 and Artemis genetic forms of SCID.40,41
Gene correction
Correction of single base mutations (and potentially bigger lesions) is possible by using endogenous DNA repair mechanisms to perform homologous recombination using added “guide” sequence (DNA or RNA/DNA). This homology-directed repair (HDR) would result in correction of the defective disease-causing gene within its normal chromosomal context for physiologic expression. Initial attempts at HDR by adding homologous donor guide sequences achieved only low efficiency of gene modification (0.01-0.1%), with poor reproducibility. Subsequently, it was discovered that the rate of site-specific HDR can be significantly increased by making a DNA double-strand break at the target sequence (1%–10%).
The most common approach being studied is a 2-step process using an engineered site-specific endonuclease to introduce a double-stranded DNA break near a genomic target and harnessing the cellular HDR DNA repair process with a homologous nucleic acid “donor” guide sequence (oligonucleotide or carried in a plasmid or viral vector) that introduces the desired mutation correction. HDR may use the donor sequence as a template to bridge the gap at the double-stranded break, copying in corrective sequence present in the donor. A succession of engineered endonucleases have been developed (homing endonucleases, zinc finger nucleases, TALENs, and CRISPR/Cas9 nucleases) to introduce the targeted double-stranded DNA break; there are passionate devotees of each, but minimal direct comparative data to define which is the most active and the most site specific. These techniques work with sufficient efficiency for gene modification in cell lines and even in induced pluripotent stem cells and others, in which correctly engineered clones may be expanded. The challenge is greater for gene correction of primary HSCs, in which a high percentage of the cells need to be corrected with minimal toxicity or loss of stem cell capacity for use in autologous transplantation. To date, the levels of gene correction have been too low for likely clinical benefit,42,43 although progress is being made.44
Summary
The outcome for infants with SCID is expected to improve as NBS identifies them before the onset of severe infections. The optimal approaches to treatment remain to be determined and there are almost as many approaches to the treatment of SCID as there are genetic forms and treating transplantation centers. Allogeneic HSCT is limited by immunological risks (GVHD) and autologous/gene therapy has risks from the genetic manipulation. New methods (reduced-intensity conditioning, improved graft engineering, safer vectors, and effective gene correction) are advancing to the clinical setting and may retain or increase the efficacy of allogeneic HSCT and autologous HSCT/gene therapy and decrease risks. SCID continues to guide the way to improving HSCT.
Republished in 2012 on the 40th anniversary of the original report.19
Acknowledgments
The NIH provided support for the author's cited research studies (Grants PO1 HL073104, U01 AI100801, U01 AI087628, and R01 FD003005). Sung-yun Pai (Boston Children's Hospital), Roger Hollis (UCLA), and Megan Hoban (UCLA) provided helpful reviews of the manuscript.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Donald B. Kohn, MD, Departments of Microbiology, Immunology and Molecular Genetics and Pediatrics, University of California, Los Angeles, 610 Charles E. Young Dr., 3163 Terasaki Life Science Bldg., Los Angeles, CA 90095; Phone: (310)794-1964; Fax: (310)206-0356; e-mail: dkohn1@mednet.ucla.edu.
