
Abstract
Sickle cell disease (SCD) is an inherited disorder secondary to a point mutation at the sixth position of the beta chain of human hemoglobin that results in the replacement of valine for glutamic acid. This recessive genetic abnormality precipitates the polymerization of the deoxygenated form of hemoglobin S that induces a major distortion of red blood cells (sickle red blood cells), which decreases sickle red blood cell deformability, leading to chronic hemolysis and vasoocclusion. These processes can result in severe complications, including chronic pain, end organ dysfunction, stroke, and early mortality. The only proven curative therapy for patients with SCD is myeloablative conditioning and allogeneic stem cell transplantation from HLA-matched sibling donors. In this review, we discuss the most recent advances in allogeneic stem cell transplantation in SCD, including more novel approaches such as reduced toxicity conditioning and the use of alternative allogeneic donors (matched unrelated donors, umbilical cord blood transplantation, haploidentical donors) and autologous gene correction stem cell strategies. Prospects are bright for new stem cell approaches for patients with SCD that will enable curative stem and genetic correction therapies for a greater number of patients suffering from this chronic and debilitating condition.
Learning Objective
To highlight the current state of the art for allogeneic transplantation and corrective genetic cellular therapy for sickle cell disease
Introduction
In 1984, Johnson et al1 first demonstrated the success of a myeloablative conditioning (MAC) and HLA-matched sibling allogeneic hematopoietic stem cell transplantation (allo-HSCT) in a patient with sickle cell disease (SCD). Walters et al2 subsequently reported on the successful use of HLA-matched sibling MAC allo-HSCT in a larger series of patients with SCD. Twenty-two patients with severe symptoms of SCD received allo-HSCT from a fully HLA-matched sibling donor (MSD) after receiving a MAC regimen consisting of busulfan (Bu), cyclophosphamide (Cy), and antithymocyte globulin (ATG).2 The event-free survival (EFS) and disease-free survival (DFS) rates at 4 years were 91% and 73%, respectively.2 Graft failure occurred in 19% of allo-HSCT SCD recipients. One patient experienced early graft rejection at day 30 and the remaining 3 patients experienced late graft rejection between days 156 and 270. In 2007, the Center for International Blood and Marrow Transplant Research (CIBMTR) reported on 67 pediatric patients who received HLA-matched sibling allo-HSCTs after a MAC regimen.3 Panepinto et al3 demonstrated that the 5-year overall survival (OS) and DFS rates were 97% and 85%, respectively. The incidence of graft failure was 15%. The majority of the graft failures occurred late and approximately one-half of the patients had >10 packed RBC transfusions before BM transplantation (BMT) as a possible predisposing risk factor for developing graft failure. The rates of acute GVHD (AGVHD, grade II-IV) and chronic GVHD (CGVHD) were 10% and 22%, respectively. Bernaudin et al4 reported the results from France demonstrating similar results in 87 allo-HSCT SCD recipients who received allo-HSCTs from HLA MSDs after MAC with Bu and Cy. The 6-year OS was 93.1% and the EFS was 86.1%, respectively. Graft failure occurred in 7% of these patients; however, after the introduction of ATG as part of the conditioning regimen, the incidence of graft failure decreased to 2.9%. Six rejections occurred at 5-100 months after transplantation. One patient failed to engraft after a matched sibling cord blood (CB) transplantation; that patient experienced autologous reconstitution despite a second BM graft from the same donor. The probability of AGVHD (grade II-IV) and CGVHD was 20% and 12.6%, respectively.4
Locatelli et al5 recently reported the results of 130 Eurocord registry patients who underwent HLA-identical sibling BMT and 30 patients who underwent HLA-identical sibling CB transplantations for SCD. All patients received conditioning that included Bu and the majority of patients who received BM also received Cy. Patients receiving CB were more likely to receive Bu combined with fludarabine (Flu) or thiotepa-based regimens. ATG was used in the majority of patients as well. The 6-year DFS for all SCD patients was 92 ± 2%. Specifically the DFS at 6 years after BMT was 92% ± 2% and after sibling CB transplantation it was 90% ± 5% (Table 1).
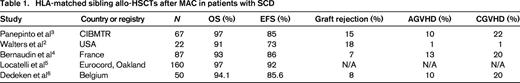
Dedeken et al6 reported the outcome of 50 consecutive children with severe SCD that received HLA-matched sibling allo-HSCT from 1988 to 2013. The stem cell source was BM (n = 39), sibling CB (n = 3), both (n = 7), and PBSCs (n = 1). The MAC regimen consisted of Bu and Cy (Bu/Cy) before November 1991 and Bu/Cy with rabbit ATG after that date. Since 1995, all patients have been treated with hydroxyurea before transplantation for a median of 2.7 years. AGVHD and CGVHD were observed in 11 and 10 patients, respectively. Eight-year OS and EFS rates were 94.1% and 85.6%, respectively. Since the introduction of hydroxyurea, no graft failures have occurred and the EFS now approaches 97.4% (Table 1).6 These studies demonstrate that HLA-matched sibling allo-HSCTs after MAC offer very high survival rates with few transplantation-related complications (Table 1).
Long-term (late effects) of HLA sibling-matched allo-HSCT and MAC in patients with symptomatic SCD
Walters et al7 reported on late effects of MSD allo-HSCT in children with severe SCD who underwent transplantation between 1991 and 2000. After allo-HSCT, patients with stroke who had stable engraftment of donor cells experienced no subsequent stroke events after BMT and brain magnetic resonance imaging examinations demonstrated stable or improved results. However, 2 patients with graft rejection had a second stroke after BMT. After transplantation, most patients also had unchanged or improved pulmonary function. There was, however, significant gonadal toxicity after BMT, particularly among female recipients. Other investigators have also demonstrated that allo-HSCT has been demonstrated to stabilize or reverse the organ damage secondary to SCD. In a long-term follow-up study of patients who received HLA-matched related allo-HSCT, pulmonary function was stable in 22 of 26 patients, worse in 2, and not studied in 2.8 Linear growth measured by median height SD score improved from −0.7 before HSCT to −0.2 after HSCT.8
Reduced-intensity conditioning and allo-HSCT in pediatric SCD recipients
A major limitation of MAC and allo-HSCT is the risk of transplantation-related mortality or treatment-related toxicities associated with MAC regimens. Organ toxicities are more likely to occur and be more severe in symptomatic patients with SCD who have impaired organ function or have been exposed to multiple RBC transfusions before MAC and allo-HSCT. MAC facilitates durable engraftment of donor cells, but is limited by conditioning related toxicities and transplantation-related complications.9 The ongoing National Heart, Lung, and Blood Institute (NHLBI) BMT clinical trials network (CTN) 0601 trial uses a reduced-intensity conditioning (RIC) regimen that includes alemtuzumab, Flu, and melphalan as a conditioning regimen, followed by an 8/8 HLA matched adult unrelated donor (MUD) transplantation. GVHD prophylaxis consists of cyclosporine/FK506, methotrexate, and steroids. However, the major difficulty with this approach is identifying an 8/8 HLA MUD because there are lower percentages of African-American and Hispanic-American donors in the international BM registries. Only ∼20%-25% of patients identified as potential BMT candidates have an 8/8 HLA MUD available. Clearly, other strategies to increase the donor pool or other alternatives are desperately needed for identifying allogeneic donors for SCD. This trial is nearing completion and the results will be forthcoming (S. Shenoy, Washington University, St. Louis, MO, personal communication).
Krishnamurti et al10 reported on stable donor engraftment after RIC with Bu, Flu, equine ATG, and total lymphoid irradiation. Six of 7 patients with SCD demonstrated long-term engraftment. Four of 6 patients had evidence of only partial donor chimerism, but these patients still had a hemoglobin level >10 and alleviation of their symptoms of SCD (Table 2).
RIC/RTC regimens before allo-HSCT in patients with symptomatic SCD
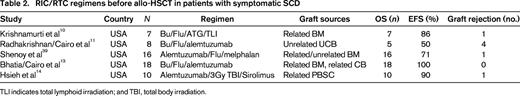
TLI indicates total lymphoid irradiation; and TBI, total body irradiation.
Radhakrishnan et al11 reported on the experience of RIC with Bu, Flu, and alemtuzumab for 8 consecutive pediatric patients with high-risk symptomatic SCD undergoing unrelated CB transplantation (UCBT). Among evaluable (5 of 8) UCBT recipients, 62.5% engrafted neutrophils at median day 34 (range, 27 to 47). In addition to the 3 unrelated UCBT recipients who did not engraft neutrophils, one unrelated UCBT recipient achieved absolute neutrophil count >500/mm3 for 3 days, but did not achieve >50% donor chimerism in whole blood by day +60 and was therefore classified as having primary graft failure. The probability of grade II to grade IV AGVHD was 50.0% and the probability of grade III to grade IV AGVHD was 25.0%. One recipient developed CGVHD, which was limited. Two-year EFS and OS were 50% and 62.5%, respectively. Three patients with primary graft failure died from infection: one died from cytomegalovirus pneumonitis on day +84; another died of adenovirus on day +128; and one patient, who had developed primary graft failure due to cytomegalovirus, received a second allograft 1 year later for persistent aplasia and died of Candida parapsilosis. The above results parallel the results reported by Kamani et al,12 with high incidence of graft failure after RIC and UCBT in pediatric recipients with high-risk SCD.
Bhatia et al13 have demonstrated the successful use of reduced-toxicity conditioning (RTC) followed by HLA-matched sibling BM or CB transplantation in patients with SCD. Eighteen patients received Bu, Flu, and alemtuzumab before receiving either matched sibling BM or CB allo-HSCT. The median age was 8.9 years (range, 2.3-20.2), and 15 sibling BM and 3 sibling CB transplantations were performed. Mean whole blood and erythroid donor chimerism were 90.7% and 87.7% at days +100 and +365, respectively. Probability of grade II-IV AGVHD was 16.7%. Two-year EFS and OS were both 100%.13 These data illustrate the well-tolerated and successful RTC regimen of Bu, Flu, and alemtuzumab in SCD patients after MSD allo-HSCT (Table 2).
Hsieh et al14 reported the results of a pilot study of nonablative conditioning and HLA-MSD allo-HSCT using 300 cGy of total body irradiation and alemtuzumab in 10 adult patients with high-risk SCD and demonstrated a 90% EFS but a 10% incidence of graft failure. However, patients required long-term immunosuppression with 50% T-cell donor chimerism at 1 year.14 This confirms that RTC can allow for sufficient donor whole blood and RBC engraftment to ameliorate SCD. These studies support the use of RIC/RTC regimens in patients with symptomatic SCD who cannot undergo a MAC allo-HSCT (Table 2).
Alternative allogeneic donor sources for allo-HSCT in patients with SCD
We and others have demonstrated that UCB is an excellent alternative allogeneic donor source for both childhood malignant and nonmalignant conditions.15-17 The preliminary results of UCBT as an alternative allogeneic source for SCD, although limited in scope, have been disappointing.11,18,19 The NHLBI BMT CTN trial of RIC UCBT in symptomatic patients with SCD was closed early to accrual secondary to increased graft rejection.12 Eight children with severe SCD underwent UCBT after alemtuzumab, Flu, and melphalan. Cyclosporine or tacrolimus and mycophenolate mofetil were administered for GVHD prophylaxis. The median pre-cryopreservation total nucleated cell dose was 6.4 × 107/kg (range, 3.1-7.6) and the median post-thaw infused CD34 cell dose was 1.5 × 105/kg (range, 0.2-2.3). Three patients engrafted had 100% donor cells by day 100, which was sustained, and 5 patients had autologous hematopoietic recovery. With a median follow-up of 1.8 years (range, 1-2.6), 7 patients are alive with a 1-year survival of 100% and 3 of 8 are alive without graft failure or disease recurrence.12
Ruggeri et al19 examined the efficacy of UCBT in children with SCD (N = 16). OS and DFS were 94% and 50%, respectively. The 2-year probability of DFS was 45% in patients who received allografts with nucleated cell dose >5 × 107/kg compared with only 13% with lower cell doses. Primary graft failure was the predominant cause of treatment failure, occurring in 7 patients with SCD. These results suggest that only UCB units containing an expected infused nucleated cell dose >5 × 107/kg should be considered for transplantation for hemoglobinopathies, which further limits the available UCB units for this population of patients.19
Bolanos-Meade et al20 reported on using a nonmyeloablative approach in 17 adult patients, 14 from HLA-haploidentical donors and 3 from HLA-matched related donors. The regimen consisted of ATG, Flu, Cy, total body irradiation, and GVHD prophylaxis with posttransplantation high-dose Cy, mycophenolate mofetil, and tacrolimus or sirolimus. Graft failure was not seen in HLA-matched patients, but 43% of the haploidentical patients rejected their graft. Only one patient developed skin-only AGVHD and there was no mortality. Therefore, non-MAC with posttransplantation high-dose Cy expands the donor pool, making BMT feasible for most patients with SCD, and is associated with a low risk of complications. However, further investigation is needed to lower the rejection rate.20
Familial haploidentical T-cell-depleted transplantation
Familial haploidentical (FHI) T-cell depletion (TCD) is another approach to overcoming the paucity of well-matched related and unrelated donors in this patient population.21 For patients with thalassemia, Lucarelli et al originally designed a conditioning regimen consisting of hydroxyurea and azathioprine between days −45 and −11 before Flu 20 mg/m2/d × 6 days, Bu 14 mg/kg/total dose and Cy 60 mg/kg/total dose in 33 poor-risk class 3 thalassemia patients before allo-HSCT. Graft rejection was reduced to only 8% compared with the previous 30% reported from the same group without the hydroxyurea, azathioprine, and Flu.22,23 Further improvements were achieved in 22 poor-risk thalassemia patients by modifying the conditioning regimen to add thiotepa 10 mg/kg/ × 1, rabbit ATG 12.5 mg/kg/total dose, expanding the use of hydroxyurea and azathioprine to day −59, and increasing Cy to 200 mg/kg/total dose. Grafts primarily from a maternal donor (N = 20) were depleted of T cells using the CliniMACS system to achieve a median of 14.2 × 106 CD34+ cells/kg with a controlled add-back of 2 × 105 CD3+/kg and cyclosporine added as AGVHD prophylaxis. Engraftment was achieved in 16/22 patients without AGVHD and with OS of 90%. These results suggest that a similar approach could be investigated in high-risk patients with SCD.
We have created a multicenter, multidisciplinary consortium of pediatric SCT centers, each having a substantial SCD patient population with the intent of investigating FHI TCD allo-HSCT in high-risk patients with SCD.21 We have adopted the conditioning regimen that Lucarelli piloted in the FHI TCD allo-HSCT study in a high-risk thalassemia population (Figure 1A). We have included the addition of total lymphoid irradiation to potentially reduce the rejection rate in this SCD population. Selected high-risk patients with SCD will be enrolled on this study. Patients will receive a myelosuppressive/immunosuppressive conditioning regimen and will receive FHI TCD (CD34 selected) and T-cell add-back PBSC transplantation using the CliniMACS device IND #14359 (Figure 1B).

Myeloimmunosuppressive conditioning regimen and cellular processing for FHI SCD clinical trial. (A) MAC regimen before haploidentical allo-SCT in high-risk SCD. (B) CD34 selection and T-cell add-back procedure.
Myeloimmunosuppressive conditioning regimen and cellular processing for FHI SCD clinical trial. (A) MAC regimen before haploidentical allo-SCT in high-risk SCD. (B) CD34 selection and T-cell add-back procedure.
Four patients have received allo-HSCTs to date. All used maternal donors without complications and had early myeloid engraftment, ≥97% whole blood and ≥96% RBC donor chimerism, and no AGVHD or CGVHD.24
Early results indicate FHI TCD allo-HSCT is feasible in high-risk SCD patients who lack an MSD or MUD.24 A larger cohort is needed to assess long-term safety and outcomes [supported by Food and Drug Administration (FDA) Grant 5R01FD004090 and a grant from Otsuka; IND #14359 and www.clinicaltrials.gov identifier NCT 01461837].
Gene therapy
Although allo-HSCT is the only proven curative option for patients with high-risk SCD, this therapeutic approach is limited by lack of HLA well-matched family and unrelated donors and allo-HSCT related morbidity [acute and chronic (late)] and mortality.21 To circumvent this limitation, gene therapy using autologous stem cells offers an alternative therapeutic strategy to overcome lack of HLA-matched allogeneic donors and allo-HSCT-related morbidity and mortality.25 Alternatively, recent investigations of developing induced pluripotent stem cells (iPSCs) and new gene-editing approaches to deliver site-specific double-stranded DNA breaks allows for both nonhomologous end joining and homology-directed repair/homologous reconstitution facilitating gene correction strategies in SCD.26
In the past, gene therapy strategies have included approaches to modify β-globin production to develop synthetic β-globins with antisickling properties and gamma globin strategies to enhance hemoglobin F (HgF) production and reduce hemoglobin S (HgS) production.25 The β-globin gene cluster is located on human chromosome 11 and consists of 5 functional genes including ε, Gγ, Aγ, δ, and β (Figure 2). In the second and third trimesters of fetal life, the γ-globin gene on chromosome 11 is predominantly expressed in the form of HgF (α2γ2) and the β-globin gene is significantly repressed. After birth, the opposite occurs and γ-globin expression is significantly repressed.25 The regulation of γ-globin to β-globin switching and β-globin expression is highly complex and is regulated by several transcription factors and histone deacetylases (HDAC1, HDAC2) and 5 locus control regions (GATA1, BCL11A, KLF-1, MYB, SOX6) highly sensitive to DNase 1 in erythroid cells 40-60 kb upstream from the β-globin gene, respectively (Figure 2).25,27,28

Human γ-globin gene locus on chromosome 11 showing the ontology of expression of the embryonic, fetal, and adult globin genes controlled by locus control regions. In adult life, the transcription of γ-globin is highly repressed. Some of the major transcription factors involved in the repression of γ-globin are highlighted. Reprinted with permission from Chandrakasan and Malik, 2014.25
Human γ-globin gene locus on chromosome 11 showing the ontology of expression of the embryonic, fetal, and adult globin genes controlled by locus control regions. In adult life, the transcription of γ-globin is highly repressed. Some of the major transcription factors involved in the repression of γ-globin are highlighted. Reprinted with permission from Chandrakasan and Malik, 2014.25
Multiple gene therapy approaches have been investigated over the past 15 years as potential options for patients with high-risk SCD. Pawliuk et al29 first demonstrated the modification of sickle β-globin production to BT87Q by a lentiviral vector (LV) in a murine model of SCD and demonstrated this genetic mutation had significant antisickling properties. Similar approaches using a Sleeping Beauty transposon (nonviral) approach via a nucleofection gene transduction approach with BT87Q in CD34+ HSCs from a patient with SCD has also been demonstrated to significantly reduce RBC sickling.30 After this initial observation with modest effects on sickling complications, Levasseur et al31 demonstrated the production of the antisickling βAS3-globin using a self-inactivating LV in sickle mice with prior expression of human α-globin and βS-globin. Most recently, Romero et al,32 using CD34 HSCs from BM of a patient with SCD, successfully induced βAS3-globin production using an LV-based vector that was associated with a reduction in HgBS associated physiological changes. A summary of gene therapy/transduction approaches to induce β-globins with antisickling properties is illustrated in Figure 3.

Gene therapy viruses and vectors. The genome of both wild-type murine MLV (A) and the HIV-1 virus and the gene therapy vectors derived from them (B). The initial retroviral vectors had full-length long terminal repeats (LTRs) with intact U3 region (which carries the viral enhancer and promoter). With the current generation of LVs, the U3 region of the 3′ LTR is deleted and in the 5′ LTR, it is replaced by a CMV promoter in the 5′ LTR. The CMV promoter is only used in packaging the vector and is not transmitted to host cells. The HIV envelope is replaced by the VSV-G envelope. (C) Prototypic globin LV vector. Reprinted with permission from Chandrakasan and Malik, 2014.25
Gene therapy viruses and vectors. The genome of both wild-type murine MLV (A) and the HIV-1 virus and the gene therapy vectors derived from them (B). The initial retroviral vectors had full-length long terminal repeats (LTRs) with intact U3 region (which carries the viral enhancer and promoter). With the current generation of LVs, the U3 region of the 3′ LTR is deleted and in the 5′ LTR, it is replaced by a CMV promoter in the 5′ LTR. The CMV promoter is only used in packaging the vector and is not transmitted to host cells. The HIV envelope is replaced by the VSV-G envelope. (C) Prototypic globin LV vector. Reprinted with permission from Chandrakasan and Malik, 2014.25
Several gene therapy approaches to enhance γ-globin production and enhance HgF production have also been investigated over the past several years.25 Pestina et al33 originally demonstrated the ability to enhance HgF production and amelioration of SCD-related symptoms by an LV-mediated transfer of the 3.1 kb of the locus control region and 130 bp β-globin promoter region. Perumbeti et al34 developed a novel γ-globin gene vector by substituting the exons for γ-globin in the β-globin gene and demonstrated 10% production of HgF and 20% donor chimerism in a humanized sickle cell mouse model. Lastly, the Orkin group demonstrated that BCL11A is a major repressor of human γ-globin expression and that silencing of BCL11A in humanized sickling mice significantly enhances HgF production and SCD-related hematological and pathological defects (Figure 2).35,36
Most recently, the technology to develop iPSCs from mature somatic cells has allowed advanced gene editing approaches using site directed endonucleases, such as zinc finger nucleases, transcription activator-like effector nucleases, and clustered regulatory interspaced short palindromic repeat endonucleases, to induce double-stranded DNA breaks and after nonhomologous end joining or homology-directed repair/homologous reconstitution gene correction strategies (Figure 4A-D).21,37,38 Future studies are required to determine the long-term efficacy and safety of these recent iPSC gene-editing strategies.

Development of SCD patients. (A) Studies deriving gene-corrected HSCs using human cell lines obtained from patients with SCD to generate iPS cells, with the ultimate goal of autologous transplantation. Reprinted with permission from Freed et al, 2012.21 (B) Sleeping Beauty (SB) transposon and transposase mediated wild-type (Wt) β-globin gene integration. (C) Transcription activator-like effector nucleases (TALENs)-mediated human beta-globin (HBB) gene correction in SCD. (D) The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas)-mediated human β-globin (HBB) gene correction in SCD.
Development of SCD patients. (A) Studies deriving gene-corrected HSCs using human cell lines obtained from patients with SCD to generate iPS cells, with the ultimate goal of autologous transplantation. Reprinted with permission from Freed et al, 2012.21 (B) Sleeping Beauty (SB) transposon and transposase mediated wild-type (Wt) β-globin gene integration. (C) Transcription activator-like effector nucleases (TALENs)-mediated human beta-globin (HBB) gene correction in SCD. (D) The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas)-mediated human β-globin (HBB) gene correction in SCD.
Currently, St Jude's Research Hospital has an open clinical trial entitled “Retroviral Vector Mediated Globin Gene Transfer to Correct Sickle Cell Anemia or Thalassemia” (www.clinicaltrials.gov identifier NCT00669305). CD34+ cells purified from the BM of research participants with a sickle cell syndrome or a thalassemia syndrome will be transduced with retroviral vectors containing γ-globin coding sequences under the control of the β-globin gene promoter and including various regulatory elements chosen to enhance gene expression and to insulate regulatory elements from cellular genes at or near the integration sites. The efficiency of gene transfer and the function of the globin transgene will be evaluated in erythroid cells derived from transduced progenitors and from the progenitors in the BM of immunodeficient mice engrafted with transduced, primitive hematopoietic cells. This study will evaluate whether a vector can be designed to achieve both a potentially therapeutic efficiency of gene transfer into repopulating cells and a potentially therapeutic level of globin gene expression in maturing erythroid cells. A second clinical trial is about to open at the time of this writing entitled “A Phase 1 Study Evaluating Gene Therapy by Transplantation of Autologous CD34+ Stem Cells Transduced Ex Vivo With the LentiGlobin BB305 Lentiviral Vector in Subjects With Severe Sickle Cell Disease.” This is a nonrandomized, open label, multisite, single-dose, phase 1 study in up to 8 adults with severe SCD. The study will evaluate the safety and efficacy of the LentiGlobin BB305 drug product consisting of autologous CD34+ HSCs transduced with LentiGlobin BB305 LV vector encoding the human beta A-T87Q-globin gene (www.clinicaltrials.gov identifier NCT02140554, sponsored by BluBird Bio).
Summary
MAC with HLA MSD allo-HSCT is the only known curative therapy in patients with SCD. More novel approaches are being investigated, including RIC and the use of alternative allogeneic donors (MUDs, UCBT, haploidentical) and autologous gene correction/replacement stem cell therapies. Prospects are bright for new stem cell approaches for patients with SCD and we are able to offer a greater number of patients a potential cure from this chronic and debilitating condition.
Acknowledgments
This work was supported in part by grants from the FDA (Grant 5R01FD004090) and the Pediatric Cancer Research Foundation. The authors thank Yaya Chu and Sanghoon Lee for their significant contribution to the production of Figure 4A-D and Erin Morris for her editorial assistance in the production of this manuscript.
Disclosures
Conflict-of-interest disclosures: M.S.C. has received research funding from Otsuka. J.T. declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Mitchell S. Cairo, MD, Chief, Pediatric Hematology, Oncology and Stem Cell Transplantation, Director, Children and Adolescent Cancer and Blood Diseases Center, Medical and Scientific Director, Cellular and Tissue Engineering Laboratory, Associate Chairman, Department of Pediatrics, Professor of Pediatrics, Medicine, Pathology, Microbiology & Immunology and Cell Biology & Anatomy, Maria Fareri Children's Hospital at Westchester Medical Center, New York Medical College, 40 Sunshine Cottage Rd, Skyline Office, 1N-D12 Valhalla, NY 10595; Phone: 914-594-2150; Fax: 914-594-2151; e-mail: mitchell_cairo@nymc.edu.
