
Abstract
The aims of this review are to highlight the mechanisms and consequences of iron distribution that are most relevant to transfused sickle cell disease (SCD) patients and to address the particular challenges in the monitoring and treatment of iron overload. In contrast to many inherited anemias, in SCD, iron overload does not occur without blood transfusion. The rate of iron loading in SCD depends on the blood transfusion regime: with simple hypertransfusion regimes, rates approximate to thalassemia major, but iron loading can be minimal with automated erythrocyte apheresis. The consequences of transfusional iron overload largely reflect the distribution of storage iron. In SCD, a lower proportion of transfused iron distributes extrahepatically and occurs later than in thalassemia major, so complications of iron overload to the heart and endocrine system are less common. We discuss the mechanisms by which these differences may be mediated. Treatment with iron chelation and monitoring of transfusional iron overload in SCD aim principally at controlling liver iron, thereby reducing the risk of cirrhosis and hepatocellular carcinoma. Monitoring of liver iron concentration pretreatment and in response to chelation can be estimated using serum ferritin, but noninvasive measurement of liver iron concentration using validated and widely available MRI techniques reduces the risk of under- or overtreatment. The optimal use of chelation regimes to achieve these goals is described.
Iron metabolism in sickle cell disease
In the absence of repeated blood transfusions, as in the healthy population, storage iron rarely exceeds 2000 mg in sickle cell disease (SCD), being present mainly in macrophages of liver, spleen, and BM. In marked contrast to other hemolytic anemias, iron deficiency is described in nontransfused SCD populations. A recent study in > 8000 individuals in India found that iron deficiency was more common in women with SCD (67%) than in those with sickle trait (26%) or healthy controls (22%).1 Therefore, only when repeated blood transfusions are given does iron overload develop in SCD. A key difference between SCD and thalassemia major (TM) is the primary site of RBC destruction: it is predominantly intra-BM in TM (ineffective erythropoiesis [IE]) and predominantly intravascular hemolysis in SCD. BM-derived factors such as growth differentiation factor 15 (GDF15), twisted gastrulation protein homolog 1 (TWSG1),2 or the recently identified erythroferone suppress hepcidin in TM, leading to increased dietary iron absorption, a mechanism that is relatively lacking in SCD. Intravascular hemolysis provides a potential mechanism for iron elimination in SCD through increased excretion of urinary3 and biliary4 iron as Hb, hemosiderin, or heme. Feline leukemia virus subgroup C receptor (FLVCR), a cellular and mitochondrial heme exporter, may contribute to heme iron elimination, because mice lacking FLVCR show hepatic iron overload as early as 5 weeks of age.4 Iron distribution may also be affected by the induction of heme oxygenase 1 (HO-1) after the uptake of heme into tissues (see “Mechanisms underlying distribution of transfusional iron in SCD”). Urinary iron loss, as hemoglobin or urinary hemosiderin, is well known in other forms of intravascular hemolysis, such as paroxysmal nocturnal hemoglobinuria. In studies performed > 2 to 3 decades ago, urinary iron was found to be increased the majority of steady-state SCD patients. This increased further during hemolytic crises, reaching up to 15 mg/d (∼ 0.2 mg Fe/kg/d, comparable to the average SCD transfusional iron load rate). Mild iron deficiency in SCD might have a beneficial effect on disease severity by diminishing hemoglobin S (HbS) synthesis in favor of fetal hemoglobin (HbF).5 This is summarized in Figure 1.

Summary of differences in iron turnover in SCD triggered by transfusion. This scheme is devised by the authors and builds upon current understandings from work by us and others.16,23,29 (A) Iron turnover in SCD in the absence of blood transfusion is shown. Arrows denote iron fluxes involved in RBC formation in the BM from plasma transferrin pool (Tf) and destruction in RES macrophages (black) or through intravascular hemolysis (grey). Intravascular hemolysis forms a significant proportion of iron turnover directed to liver via hemopexin and haptoglobin binding of heme and hemoglobin, respectively. These mechanisms are nearly always saturated in SCD with a large proportion of free Hb available for glomerular filtration and renal uptake (via megalin and cubulin) leading to renal iron redistribution and urinary loss. IE in SCD is small relative to TM and effective erythropoiesis in SCD, so that BM is less expanded and IE iron reflux smaller than in TM, whereas iron absorption from the gut is not as increased through suppression of hepcidin. Hepatocytes and RES have low iron stores despite high rate of iron entry (as heme, Hb, or RBC) due to unopposed iron exit via ferroportin secondary to low hepcidin50 and to heme-dependent up-regulation of ferroportin transcription.32 Hypoxia and high rate of erythropoiesis reduce hepcidin51 despite chronic inflammatory state,52 leading to relative hepcidin deficiency (low hepcidin/ferritin ratios) that facilitates iron egress from RES and hepatocytes. Urinary loss may be greater than intestinal absorption, leading to iron deficiency in a high proportion of nontransfused SCD patients. (B) Iron turnover in a transfused SCD patient is shown. Replacement of sickle RBCs with transfused RBCs decreases intravascular hemolysis (grey) and hence decreases iron clearance through hemopexin and haptoglobin by the liver and leaves less free Hb available for kidney uptake and urinary loss. Erythropoiesis of sickle RBCs is also suppressed if that transfusion regime increases the Hb. A greater proportion of iron turnover is through extravascular RBC destruction, which is subsequently directed via Tf to BM and hepatocytes. Increased Hb values after transfusion decrease erythropoiesis and transferrin iron clearance in BM: a greater proportion of transferrin iron is directed to hepatocytes which store increased iron (shown in dark grey). RES iron is also increased by greater erythrophagocytosis of transfused RBCs and increased hepcidin synthesis in hepatocytes (less hypoxia and lower erythropoietic rate). Tf saturation increases relative to nontransfused SCD, but rarely to the levels seen in TM and typically without NTBI formation. Continued hemolysis of remaining sickle RBCs continues to route heme iron away from the erythrophagocytosis-transferrin circuit to hepatocyte and kidney so that Tf saturation does not increase as much as in nonhemolytic conditions despite reduced erythroid uptake.
Summary of differences in iron turnover in SCD triggered by transfusion. This scheme is devised by the authors and builds upon current understandings from work by us and others.16,23,29 (A) Iron turnover in SCD in the absence of blood transfusion is shown. Arrows denote iron fluxes involved in RBC formation in the BM from plasma transferrin pool (Tf) and destruction in RES macrophages (black) or through intravascular hemolysis (grey). Intravascular hemolysis forms a significant proportion of iron turnover directed to liver via hemopexin and haptoglobin binding of heme and hemoglobin, respectively. These mechanisms are nearly always saturated in SCD with a large proportion of free Hb available for glomerular filtration and renal uptake (via megalin and cubulin) leading to renal iron redistribution and urinary loss. IE in SCD is small relative to TM and effective erythropoiesis in SCD, so that BM is less expanded and IE iron reflux smaller than in TM, whereas iron absorption from the gut is not as increased through suppression of hepcidin. Hepatocytes and RES have low iron stores despite high rate of iron entry (as heme, Hb, or RBC) due to unopposed iron exit via ferroportin secondary to low hepcidin50 and to heme-dependent up-regulation of ferroportin transcription.32 Hypoxia and high rate of erythropoiesis reduce hepcidin51 despite chronic inflammatory state,52 leading to relative hepcidin deficiency (low hepcidin/ferritin ratios) that facilitates iron egress from RES and hepatocytes. Urinary loss may be greater than intestinal absorption, leading to iron deficiency in a high proportion of nontransfused SCD patients. (B) Iron turnover in a transfused SCD patient is shown. Replacement of sickle RBCs with transfused RBCs decreases intravascular hemolysis (grey) and hence decreases iron clearance through hemopexin and haptoglobin by the liver and leaves less free Hb available for kidney uptake and urinary loss. Erythropoiesis of sickle RBCs is also suppressed if that transfusion regime increases the Hb. A greater proportion of iron turnover is through extravascular RBC destruction, which is subsequently directed via Tf to BM and hepatocytes. Increased Hb values after transfusion decrease erythropoiesis and transferrin iron clearance in BM: a greater proportion of transferrin iron is directed to hepatocytes which store increased iron (shown in dark grey). RES iron is also increased by greater erythrophagocytosis of transfused RBCs and increased hepcidin synthesis in hepatocytes (less hypoxia and lower erythropoietic rate). Tf saturation increases relative to nontransfused SCD, but rarely to the levels seen in TM and typically without NTBI formation. Continued hemolysis of remaining sickle RBCs continues to route heme iron away from the erythrophagocytosis-transferrin circuit to hepatocyte and kidney so that Tf saturation does not increase as much as in nonhemolytic conditions despite reduced erythroid uptake.
Impact of transfusion regime on iron accumulation in SCD
The age of commencing blood transfusion, the rate of blood transfusion, and the nature of the transfusion regime itself all affect the rate and extent of iron overload in SCD.6 Historically, blood transfusions in SCD were sporadic, given by top-up transfusion or by some form of exchange procedure in response to acute episodes. With the emergence of data from the STOP trials for preventing primary and secondary stoke in SCD, a higher proportion of patients, perhaps 15%,7 will begin regular transfusion to prevent stroke in childhood. This, together with a wider use of transfusion to prevent or treat other complications, such as chest syndrome or in preparation for major surgery, puts an increasing proportion of patients at risk of iron overload. For example, the proportion of adult patients who had received blood transfusion increased from 15% in 2000 to 19% in 2009 in one major center in the United Kingdom.8 Repeated simple blood transfusions will inevitably lead to overload when the rate exceeds the losses in urine and feces. A unit of RBCs, processed from 420 mL of donor blood, contains ∼ 200 mg of iron (0.47 mg iron/mL of whole donor blood or 1.08 mg iron/mL of pure RBCs).9 The iron-loading rate depends on whether top-up or exchange transfusions are used, the type of exchange (manual or automated), the target Hb before and after exchange, and the target %HbS after exchange. The mean iron-loading rate in 195 patients receiving a variety of transfusion regimes was 0.22 mg/kg/d,10 approximately one-half of that reported in a similar study in TM (0.4 mg/kg/d).11 SCD patients who received exchange transfusions had a greater reduction in liver iron with chelation therapy than those receiving simple transfusions.10 The iron accumulation rates with 50%HbS targets using simple transfusion or with automated erythrocyte apheresis are summarized in Table 1.12 The net iron balance with automated erythrocyte apheresis also depends on the intrinsic RBC production: subjects with pre-erythrocyte apheresis Hb levels > 8.0 g/dL had lower iron accumulation and less donor blood usage than subjects with Hb levels ≤ 8.0 g/dL.12 In our own practice, normalization of serum ferritin (SF) in patients with previous severe iron overload has been achieved with continuous erythrocyte apheresis with target levels of HbS < 30% before exchange and 10% after exchange, combined with the use of deferasirox (DFX; Figure 3). Venous access can be limiting because 2 good peripheral veins are required to achieve acceptable flow rates; otherwise, repeated femoral access may be necessary, sometimes leading to scarring. Vortex ports are another option, but carry a risk of infection or thrombosis. Manual exchanges are often performed when automated erythrocyte apheresis is unavailable, leading to an intermediate iron-loading rate. Lower flow rates are required than in automated procedures, but only ∼ 30% of total blood volume can be exchanged at a time. In adults, if 4 units are removed and 3 replaced with the difference in volume made up with saline, a net Hb increase of ∼ 1 g/dL occurs due to the greater hematocrit of the transfused than the venesected blood. Net iron loading estimated from ferritin increments is ∼ 40% of that obtained with simple top-up transfusion of one unit.9
Iron balance with transfusion regimes in SCD and chelation doses required to balance iron loading

The rate of iron loading varies considerably with the transfusion modality used. The loading rate with simple transfusion is higher when the target %HbS is lower (Kim et al12 ). Automated exchange can be adjusted to achieve minimal iron loading. Iron excretion as a function of dose has been calculated for DFO and DFX chelation therapy (Cohen et al11 ) so that appropriate doses to maintain iron balance can be shown. Larger doses are required to decrease SF or LIC.

Labile plasma iron is shown across disease states in patients groups with broadly similar SF values. It can be seen that LPI is lower in SCD than other transfusion-dependent anaemias. Adapted with permission from Porter et al.53
Labile plasma iron is shown across disease states in patients groups with broadly similar SF values. It can be seen that LPI is lower in SCD than other transfusion-dependent anaemias. Adapted with permission from Porter et al.53
Frequency of iron overload in SCD and its effects on clinical outcomes
Transfusional iron overload is a significant clinical issue in SCD. Iron overload was present in approximately one-third of 141 adult SCD patients at post mortem (mean age, 36 y),13 and 7% of deaths were judged to be related to iron overload.13 In a cohort of 387 young adults from Atlanta, there were 22 deaths, 45% related to iron overload (cirrhosis: 8, heart failure: 2).14 Frequency of hospital admission increased with increasing SF in 199 transfused SCD patients, although the extent to which iron overload was a cause or a consequence of disease severity was not clear.15 Within the transfused SCD group, those who died had begun transfusion and chelation therapy later compared with those who survived.
Oxidative stress and inflammation in SCD
The proinflammatory effects of iron overload are well recognized through mechanisms involving redox cycling of unliganded iron in plasma non-transferrin-bound iron (NTBI) or within cells (labile iron pools), generating hydroxyl radicals and oxidative damage to organelles, cell membranes, and DNA (for detailed review, see Kwiatkowski and Porter9 ). The proinflammatory state in SCD in iron overload has also been described and contrasted with that of TM.16 Malondialdehyde was less elevated in SCD than in TM, as was NTBI, whereas IL-6, IL–5, and IL-10 were more elevated in SCD than in TM. Gamma-tocopherol, a nitric oxide–selective antioxidant, was increased 36% in SCD relative to TM. Markers of oxidative stress such as plasma malondialdehyde and nitrate were higher in iron overloaded than nonloaded SCD patients and correlated with SF.17 Markers of oxidative stress in SCD are also significantly associated with impairment of the glutathione system.18 Oxidative stress, estimated by protein carbonyl and decreased total sulfhydryl levels, were increased in SCD patients compared with control subjects and correlated with plasma iron.19 Oxidative stress also occurs though heme-induced endothelial toxicity after intravascular hemolysis. In mouse models of SCD and TM, heme promoted oxidative stress but hemopexin decreased it. Hemopexin appeared to act by decreasing heme-iron loading in the cardiovascular system, thereby decreasing oxidative stress in the endothelium with decreased induction of adhesion molecules, and was associated with promotion of heme recovery and detoxification by the liver, mainly through the induction of HO-1 activity.20
Distribution of transfusional iron in SCD
Extrahepatic iron distribution is relatively uncommon in SCD compared with TM. Iron derived from transfused RBCs initially accumulates in macrophages (capacity of the reticuloendothelial system [RES] for storage iron), but later accumulates in hepatocytes. This was confirmed in a study of cellular iron distribution by liver biopsy in which liver iron concentration (LIC) levels < 7 mg/g dry weight iron deposition was predominantly in Kupffer cells (sinusoidal compartment) and hepatocellular iron scores in SCD only approached those of the sinusoidal compartment when total liver iron levels were high (> 15 mg/g dry weight).21 In advanced transfusional iron overload, iron deposition has also been reported in cardiomyocytes post mortem in ∼10% of patients13 and in a similar proportion of adults with advanced overload by T2* MRI occurring later in SCD than in TM. Endocrine disturbances attributable to iron overload are rare in SCD22 but MRI data are scarce. In TM, the anterior pituitary is the first part of the endocrine system to be affected by iron overload. In a preliminary study23 MRI evidence of pituitary iron was compared between controls and patients with SCD, Diamond Blackfan anemia, and TM. Although, as expected, pituitary iron was highest in Diamond Blackfan anemia and TM, there was evidence of increased pituitary iron in some heavily transfused SCD in patients with high LIC concentrations. An inverse correlation between pituitary volume and estimated pituitary iron and thus its endocrine reserve was also found. Therefore, MRI may in principle identify early pituitary iron deposition in SCD before clinical manifestations are apparent.23 An increased MRI iron signal has also been identified in the kidney24 : the signal was highest in nontransfused patients with high lactate dehydrogenase levels, lacked correlation with LIC, and was higher than in TM patients. This suggests that kidney R2* may be a biomarker for chronic hemolysis-mediated vascular complications in SCD.
Hepatic consequences of transfusional iron overload in SCD
Studies of liver biopsies in SCD have linked transfusional iron load with LIC, fibrosis, and cirrhosis.25 If transfusion is given without chelation, portal fibrosis can develop as early as 2 years after transfusion. With sequential biopsies, increased fibrosis was found in 1/3 of patients at LIC values > 9 mg/g dry weight and in direct proportion to the LIC.25 Another study of transfused SCD children showed that patients with liver fibrosis or inflammation at biopsy had higher mean LIC values of 28 mg/g dry weight than those without these complications (17.6 mg/g dry weight).26 The long-term consequences of uncontrolled liver iron is cirrhosis, but the true frequency in multitransfused adult SCD patients is not known. Post mortem studies have also found cirrhosis in 11% of all patients and in ∼1/2 of patients who died with severe liver siderosis.13 In such adult patients, the risk of liver cancer exists and it would be advisable to perform yearly abdominal ultrasound and alpha fetoprotein.
Extrahepatic consequences of transfusional iron overload in SCD
Several studies show that extrahepatic consequences, particularly endocrine and cardiac effects of iron overload, are lower or more delayed in SCD than in TM. When SCD and TM patients were matched for LIC, the incidence of cardiomyopathy and endocrine disturbances such as hypothyroidism, gonadal failure, and growth delay were less frequent below the age of 20 years in SCD than in TM. Because transfusion and clinical consequences of iron overload typically begin later in SCD than in TM, the lower effects on growth and sexual development might be attributed to the lower duration of exposure to iron overload rather than to absolute levels at a particular time point.22 However, SCD patients are unlikely to be completely protected from the extrahepatic effects of iron overload. In one study, patients with the lowest bone mass also had the highest serum iron values, although SF was within normal limits in these patients.27
Mechanisms underlying distribution of transfusional iron in SCD
SCD patients typically begin transfusion later than TM patients and the transfusion rate is typically lower, but the extent to which these or other factors are key to the low prevalence of extrahepatic iron distribution in SCD is not clear. However, myocardial iron deposition has been shown to occur after more years of transfusion in SCD than in TM.28 At similar levels of iron load, transferrin saturations and plasma NTBI levels are lower in SCD than in TM,29 which could account for decreased iron distribution to the heart or endocrine system, because NTBI is considered to be a key conduit through which iron is delivered to these tissues in iron overload. However, the mechanism by which transferrin saturation and NTBI is suppressed in SCD relative to TM at similar levels of iron overload is unclear. The role of hepcidin has been explored by the Multicenter Study of Iron Overload (MSCIO) group16 : because hepcidin transcription is up-regulated by the proinflammatory cytokine IL-6, and because a chronic inflammatory state with raised IL-6 and IL-10 levels is present in SCD,29 elevated hepcidin synthesis relative to iron overload may decrease transferrin saturation and NTBI by decreasing macrophage ferroportin. Conversely, high levels of IE in TM syndromes may decrease hepcidin synthesis. However, we have found that although plasma hepcidin levels increase with SF in transfused patients, when adjusted for SF, they do not differ from non-transfusion-dependent TM or TM, and the hepcidin to ferritin ratios are low relative to healthy controls. Other investigators have also found low hepcidin in SCD compared with healthy controls (Figure 1A). These findings make raised hepcidin synthesis an unlikely mechanism for low NTBI in SCD. Other mechanisms involving hepcidin, such as autocrine hepcidin synthesis30 or variability in ferroportin sensitivity to hepcidin,31 are being explored. Iron distribution may also be affected by the high intravascular hemolysis in SCD, which may divert iron from plasma compartment, with consequent Hb uptake and iron loss through the kidneys or hemin uptake by the liver (Figure 2). Reduction in the iron available for return to plasma from RES may lower transferrin saturation. This contrasts with TM, in which ferrokinetic studies have shown that IE redirects the majority of the erythroid iron flux (85%-95%) back to plasma. Intravascular hemolysis also induces HO-1 in the tissues to which heme is cleared. Plasma heme is elevated in SCD relative to controls, leading to higher HO-1 expression in circulating endothelial cells and potentially other cell types. When HO-1 is induced after erythrophagocytosis in macrophages, ferroportin expression (haem-BACH-1 interaction–dependent32 and iron-IRP-IRE–dependent synthesis) are increased and ferritin expression is also increased.33 The balance of these opposing effects on iron release is likely to vary with the tissues concerned.34 The 3-fold higher levels of HO-1 mRNA in SCD than in TM reported35 from liver biopsies may increase hepatocellular ferritin iron storage of iron derived from heme in SCD. Finally, ischemia and ischemic reperfusion injury modulate HO-1 expression in sickle mice, which in principle may further perturb iron retention and release.36
Assessment and monitoring of iron overload in SCD
Value of SF
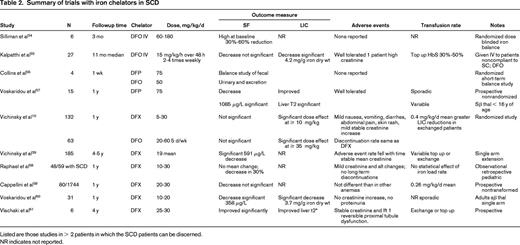
SF is the most frequently used test to estimate iron overload, but has some particular limitations in SCD. Ferritin is disproportionately increased in relation to iron loading for several weeks after a vasoocclusive sickle crisis.9 and may account for the highly variable correlations with LIC reported in the literature. If measured in steady state, several studies show a significant correlation between total transfused units by simple top-up and SF.9,37 However, aspects of this relationship are qualitatively and quantitatively nonlinear: SF is typically secreted in its iron free glycosylated form in macrophages of the RES, but secretion is finite, plateauing after ∼ 100 units of transfused blood in TM. At high body iron loads, increasing proportions of SF are derived from leakage of iron-rich unglycosylated ferritin from hepatocytes and are associated with increasing liver enzyme leakage. In SCD, sequential studies of transfused patients show that linearity exists up to SF values of ∼ 1500 to 2000 μg/L, or 20 blood transfusions, or LIC values up to 10 mg/g dry weight, but SF increases more slowly with iron load above this level.38 The ability of SF to predict LIC is best up to 10 mg/g dry weight or very high LIC values > 30 mg/g dry weight, but has relatively poor predictability between these values.38 The limitations of using SF to monitor response to chelation therapy are illustrated by a prospective randomized study comparing changes in LIC with SF changes in patients treated with DFX or deferoxamine (DFO): although a progressive and significant decrease in LIC was seen, a significant change in SF was not seen at 1 year and only became significant at 5 years of follow-up.39 With deferiprone (DFP), although there are relatively few data in SCD, the greater tropism for RES than hepatocellular iron (in contrast to DFX) as shown in TM patients, means that ferritin falls more rapidly relative to LIC than with DFX40 (Table 2). These issues put a significant constraint in the clinic when decisions are required to determine whether a patient is responding to the current chelation regime or if treatment need to be intensified or changed.
Value of LIC monitoring
The inconvenience, discomfort, and potential complications of liver biopsy can be avoided using MRI techniques. A standardized and validated MRI method is now registered in Europe and the United States (Ferriscan), with reproducible relationship between the value (R2) by MRI and LIC by biopsy over a clinically useful range41 in which locally acquired data are analyzed at a central facility. This is potentially available in any hospital with an MRI scanner and with minimal training of local staff. Other MRI techniques include T2* or R2* (1/T2*). Although the original T2* calibration underestimated LIC by a factor of 2,42 more recent calibrations43 are in closer agreement with the Ferriscan method. Other noninvasive methods for estimating LIC have been developed but are not widely available (eg, SQUID). Reliable noninvasive liver iron measurement is valuable when starting and planning chelation therapy. A good approximation to total body iron stores can be made by applying the Angelucci formula (body iron stores in mg/kg = 10.6 × the LIC in mg/g dry weight) to the measured LIC. In the absence of ongoing transfusion, the duration of chelation required to normalize iron stores can be estimated from the known iron excretion rate of the chelators in question.11 LIC measurement is also valuable when evaluating responses to a chelation regime. If there is a clear downward trend in SF, LIC estimation may not be required, but when such a trend is lacking, it is useful to decide whether the chelation regime needs modifying by assessing LIC trends.
Other monitoring of iron overload in SCD
Although myocardial iron deposition is relatively rare in SCD compared with TM, if a patient presents with a long-standing high SF or high LIC values, it is useful to know whether myocardial iron has accumulated by T2*. If this is normal, then repeat mT2* measurement is unlikely to be informative. Plasma markers of iron overload (such as NTBI or labile plasma iron) have been useful in the research setting but their role in routine monitoring has yet to be defined. Monitoring for the consequences of iron overload is also important, even though these are less frequent in SCD than in TM. This includes endocrine assessment for diabetes and hypothyroidism, as well as growth and fertility. The assessment for cirrhosis is becoming less frequent because of the decreased use of liver biopsy for iron determination. Judged by the post mortem data described above, the true frequency of cirrhosis is likely to be underrecognized. There is a need to develop validated noninvasive methodology for assessing advanced fibrosis or cirrhosis, for example using Fibroscan combined with blood markers.
Novel therapeutic strategies relating to iron and heme metabolism in SCD
Modulation of heme metabolism offers potentially useful therapeutic interventions in SCD. Iron chelation and hydroxyurea have been shown to improve oxidative status in SCD and TM.44 Vascular complications arising from release of hemoglobin or heme in circulation may also be decreased by administration of haptoglobin as shown in humans, or in animal models by hemopexin and gene therapy with HO-1. Although counterintuitive, hemin, being a potent inducer of HO-1, may have a beneficial effect when used in short priming pulses (Table 2). HO-1 activity products have also been tested for their potential benefits in IR injury and vascular pathologies including SCD. In an SCD mouse model and in in vitro studies, low levels of carbon monoxide, either inhaled or as CO-releasing molecules, were shown to suppress Hb- and hemin-dependent insults, likely by the ability of CO ability, among other things, to bind to free Hb and so render it oxidatively inert or by stimulation of HO-1 expression. Similar results in vitro were obtained with biliverdin treatment. Use of NO and NO-related products (arginine, nitrate, sildenafil), endothelin receptor blockers, Gardos channel blockers, apoA-I-mimetic peptides, and niacin have been briefly referred to in Table 2.
Objectives and indications for chelation therapy in SCD
The aims of chelation therapy in SCD are similar to other forms of transfusional iron overload, namely to maintain body iron in tissues susceptible to iron-mediated damage at levels at which damage will not occur. Guidelines for the use of chelation therapy in SCD have been broadly based on the same principles as those for TM, with the implicit acceptance that the principles of control of iron overload are the same, both with respect to the risks of undertreating iron overload and to overchelating the patient. These recommend commencement of chelation therapy when SF is > 1000 μg/L or LIC is > 7 mg/g dry weight. National Institutes of Health guidelines include cumulative transfusions of > 120 cc of packed RBCs/kg as an additional criterion for commencing chelation therapy. As discussed under “Extrahepatic onsequences of transfusional iron overload in SCD,” the hepatic iron deposition is more common than extrahepatic spread in SCD, so a key aim of chelation in SCD is to prevent long-term liver damage. The risk of extrahepatic damage is not absent, however, so a secondary objective is to prevent the spread of iron outside the liver. Some investigators have questioned the use of chelation in SCD45 on the basis that clinical trials have focused on surrogate end points such as SF or LIC rather than survival or end organ damage such as cirrhosis. The same investigators call for “larger randomized clinical trials,” but to test end points from iron overload such as death or cirrhosis between chelated and nonchelated patients would take > 2 decades and randomization would be impractical and arguably unethical. The greater heterogeneity of SCD and its management compared with TM would add further difficulties. There is no reason to assume either from first principles or from clinical observation that cirrhosis is any less likely from iron overload in SCD than in TM patients. Although current guidelines are thus broadly reasonable, systematic data on the frequency and severity of long-term liver complications in SCD patients who have been iron-overloaded for many years with or without chelation therapy would be valuable.
Chelators currently available for treatment
Three iron chelators have been licensed in the United States and Europe for the treatment of iron overload and 2 of these, DFO and DFX, have been licensed for the treatment of iron overload in SCD. The general pharmacology, mechanisms of action, and tolerability of these chelators have been described in detail elsewhere.9 The discussion below will focus on the applicability and evidence for their use in SCD.
DFO has been available since the late 1970s, but evaluation in SCD has been hampered by paucity prospective of trials, poor compliance, or inconsistencies in treatment and monitoring policies. A retrospective analysis of US health insurance claims, including 106 SCD patients, found DFO utilization data suggesting that the majority of patients are significantly undertreated for iron overload compared with current guidelines.46 Although ∼ 200 patients with SCD have been treated in clinical studies with DFO (Table 2), most of the trials have been small, investigator led, and nonrandomized. The accumulated experience suggests that DFO acts on similar iron pools and has similar rates of iron excretion in SCD as TM. Data obtained in 62 SCD patients from a randomized trial of DFO versus DFX comparing LIC and ferritin trends and using variable dosing based on LIC10 have established the doses necessary to obtain iron balance.11 This is summarized in Table 1 for SCD. Sadly, many patients have accumulated high levels of iron before chelation is effectively introduced so that substantial negative balance is initially required. Poor adherence to subcutaneous therapy is a serious issue with DFO and some investigators have tried high-dose IV therapy to circumvent this problem (Table 2), although the safety of this approach has not been evaluated prospectively. Tolerability issues from overchelation, such as audiometric and retinal toxicity, are rare, being essentially those established with TM. There have been case reports of retinal toxicity with DFO at doses usually considered safe with TM. Overchelation (> 40 mg/kg/d) can also result in decreased growth in children with SCD. Very high doses of DFO should be avoided and have been associated with lung toxicity, which could in principle be mistaken for chest syndrome.
DFX has been developed relatively recently, so a greater number of patients have been assessed in formal trials (> 300 patients, Table 2) than was the case historically with DFO. Since the introduction of this drug in 2007, it has become the most frequently used chelator in SCD in major centers in Europe.47 The drug is given once daily as a tablet dispersed in water or fruit juice. Prospective randomized data with DFO as the comparator has been obtained in 132 patients with prospective follow-up of 5 years in 63 patients. Significant decrements in LIC were seen at 1 year and significant decrement in SF at 5 years, the mean dose increasing after 1 year. From the known transfusional iron loading and the change in LIC, the dose required to obtain iron balance depending on the iron-loading rate has been calculated and summarized in Table 1. In studies with DFX, the rate of iron loading was not calculated in some cases and variability in response between studies is likely to be explained in part by this variability. Early studies also used conservative dosing starting at 10 mg/kg, and higher doses of 20 mg/kg are usually necessary to achieve negative balance. SF is a less sensitive maker of response than LIC (Table 2), so serial LIC determination is recommended before a patient is deemed a “nonresponder.” The combination of exchange transfusion with DFX can be particularly effective in bringing iron stores to target levels (Figure 3). Although labeling suggests ceasing DFX therapy when SF reaches 500 μg/L, in practice, lower levels can be achieved provided downward dose titration is practiced to achieve a “soft landing” (Figure 3). Tolerability in SCD has been similar to TM, with mild to moderate gastrointestinal effects being the most frequent. Mild stable increases in creatinine have been observed in approximately one-third of patients. Discontinuation rates from DFX (11.4%) and DFO (11.1%) were similar. A Cochrane review before the publication of the 5-year follow-up study concluded that “deferasirox appears to be as effective as deferoxamine. However, only limited evidence is available assessing the efficacy regarding patient-important outcomes.” The short-term safety of DFX seems to be acceptable48 and, although the 5-year data are reassuring, some issues remain. Renal disease is common in SCD and although DFX has a reasonable safety margin, ongoing close monitoring of renal function is required. Because proteinuria is seen in 26% of adult patients with SCD (68% have microalbuminuria) and proteinuria may occur with DFX, it may not be clear whether such SCD patients should receive DFX. It is also not clear whether or how DFX can be given to SCD patients with mild to moderate renal impairment.

Effect of switching to automated exchanges and use of DFX in iron load in SCD for 2 patients treated at University College London Hospitals. (A) The effect of changing from manual exchanges (light shading) to automated RBC exchange (darker shading) is shown in an adult patient with SCD who remained on DFX throughout the period of observation until SF values approached normal levels. The %HbS is maintained < 40% more consistently with automated exchanges. Introduction of DFX lowers SF while on manual exchanges and SF values close to normal ranges are achieved after switching to automated exchanges. (B) Effect on SF of switching from DFO to DFX is shown, followed by the effect of switching from manual (light shading) to automated exchanges (darker shading). SF remained high (> 4000 μg/L) with high LIC (25.8 mg/g dry weight) after years of DFO and manual exchanges. The trend in SF decreases toward the normal range only after introduction of DFX and automated exchange.
Effect of switching to automated exchanges and use of DFX in iron load in SCD for 2 patients treated at University College London Hospitals. (A) The effect of changing from manual exchanges (light shading) to automated RBC exchange (darker shading) is shown in an adult patient with SCD who remained on DFX throughout the period of observation until SF values approached normal levels. The %HbS is maintained < 40% more consistently with automated exchanges. Introduction of DFX lowers SF while on manual exchanges and SF values close to normal ranges are achieved after switching to automated exchanges. (B) Effect on SF of switching from DFO to DFX is shown, followed by the effect of switching from manual (light shading) to automated exchanges (darker shading). SF remained high (> 4000 μg/L) with high LIC (25.8 mg/g dry weight) after years of DFO and manual exchanges. The trend in SF decreases toward the normal range only after introduction of DFX and automated exchange.
DFP has not been licensed specifically for the treatment of iron overload in SCD. Total experience is limited to < 50 patients; the largest study included only 23 patients (Table 2). These studies have insufficient numbers and information about blood transfusion rates to draw clear conclusions about efficacy, tolerability, and the most appropriate dose of this chelator for different transfusion regimens specifically in SCD, although no new tolerability issues were reported. The drug is usually given 3 times daily in tablet form, although a syrup form has been developed recently.49 Agranulocytosis is the most serious unwanted effect of this drug, occurring in ∼1% of TM patients so that weekly blood count monitoring is recommended. Cardiac iron overload is rare in SCD, so prescription for this purpose alone is seldom required in SCD. There are very limited data on combinations of DFP and DFO in SCD.
Summary and recommendations
Knowledge about the long-term consequences of iron overload and the benefits of treatment has been gained largely from experience with TM. Because clear differences exist between SCD and other forms of transfusional iron overload with respect to extrahepatic iron distribution, more long-term information about the risks of iron overload and the benefits of treatment are required in SCD. A greater understanding of the underlying mechanisms for these differences would also be helpful in developing strategies to minimize iron-mediated toxicity. Greater focus on the long-term hepatic consequences of iron overload is also recommended in SCD. In monitoring of iron overload in SCD, the use of SF alone is particularly problematic and MRI monitoring of LIC is recommended to minimize over- or underchelation. Short- and medium-term response to chelation in SCD is affected by the transfusion regime, and a wider use of exchange rather than top-up transfusions will decrease the demands on chelation therapy in maintaining iron overload at safe levels.
Disclosures
Conflict-of-interest disclosure: J.P. has received research funding from Novartis; has consulted for Novartis, Shire, and Celgene; and has received honoraria from Novartis and Celgene. M.G. declares no competing financial interests. Off-label drug use: licensed iron-chelating agents.
Correspondence
John Porter, MA, MD, FRCP, FRCPath, Department of Haematology, University College London, Gower Street, London WC1E 6BT, United Kingdom; Phone: ++ 44 2076796224; Fax: ++ 44 2076796222; e-mail: j.porter@ucl.ac.uk.
