
Abstract
Two of the major complications that limit the efficacy of allogeneic hematopoietic cell transplantation (allo-HCT) are disease relapse and GVHD. Due to their rapid recovery early after allo-HCT and their ability to kill malignant targets without prior exposure, natural killer (NK) cells have been considered one of the main effector cells that mediate early GVL reactions. Conversely, regulatory T ells (Tregs) have proven to be critical in facilitating self-tolerance. Both murine and human studies have demonstrated a significant role for Tregs in the modulation of GVHD after allo-HCT. This article reviews the mechanisms of how these 2 cell types carry out these functions, focusing on the post-allo-HCT period. Surprisingly, relatively few studies have addressed how Tregs and NK cells interact with one another and whether these interactions are antagonistic. Although preclinical studies suggest active cross-talk between NK cells and Tregs, early clinical studies have not shown a detrimental impact of Treg therapy on relapse. Despite this, interruption of tolerogenic signals may enhance the efficacy of NK effector functions. Methods to transiently impair Treg functions and augment NK cell alloreactivity will be discussed.
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HCT) is uniquely curative for a variety of hematological malignancies in patients who are at high risk for relapse or are refractory to conventional chemotherapy. Allo-HCT leads to leukemia eradication through both the pretransplantation preparative regimen and the posttransplantation GVL responses. The exact contribution of each to leukemia eradication is unknown, but numerous studies support the concept that GVL reactions account for a significant proportion of the antileukemia activity of allo-HCT. The cells that mediate GVL in humans have not been clearly defined, but the rapid recovery of natural killer (NK) cells after transplantation has led some investigators to focus on their role in GVL reactions. Although donor NK cells mediate antihost reactions that translate into GVL, regulatory T cells (Tregs) are also present in both the recipient and the allo-HCT graft and function to induce tolerance. Although Tregs are quite beneficial in protecting from GVHD, they also have the potential to suppress alloreactive effector responses such as those associated with GVL. Given that relapse and GVHD are 2 of the major obstacles that impede successful outcomes after allo-HCT, understanding and manipulating these 2 cell populations may have a significant impact on transplantation outcomes. Although detailed mechanistic biological paradigms for both NK cells and Tregs have been established in murine models, the purpose of this review is to discuss and highlight the interactions between human NK cells and Tregs in the setting of allo-HCT. In addition, recent advances in the clinical use of these cell populations are discussed with a focus on how they interact with one another to highlight potential opportunities to prevent either disease recurrence or GVHD after allo-HCT.
NK cells: development and function
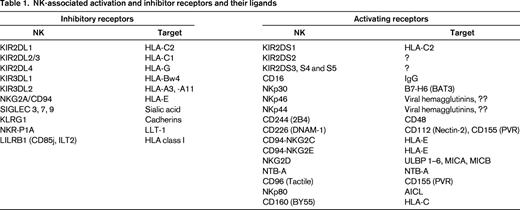
NK cells are phenotypically defined as CD3−CD56+ cells and account for ∼ 5% to 10% of the peripheral blood lymphocyte population. NK cells develop from common lymphoid progenitors that emerge from the BM and seed the secondary lymphoid tissues (lymph nodes). There, they proceed through a series of developmental intermediates under the influence of instructive cytokines (ie, IL-15).1,2 Mature NK cells are then released into the peripheral circulation, where they can rapidly produce inflammatory cytokines (ie, IFN-γ and TNF-α) or mediate cytotoxicity in response to virally infected or malignant cells. In support of this, rare individuals who lack or have dysfunctional NK cells suffer from repeated herpes or papillomavirus infections3 and some are at risk for hematopoietic malignancies.4 These 2 situations (infection or oncogenesis) both result in the reduction in surface MHC class I molecules and/or the increase in other surface molecules that are associated with cell distress. Accordingly, NK cells display a variety of both inhibitory and activating receptors that recognize MHC class I and/or stress-associated molecules. These receptors are diverse and have been outlined in previous reviews.5-7 A full listing of their names and functions is beyond the scope of this review, but some are listed in Table 1.
NK cells use their receptors to assess the status of a potential target (health or distress). In a simplified view of this, healthy cells display sufficient MHC class I and have no expression of distress molecules. In contrast, upon viral infection or malignant transformation, cells down-modulate MHC class I and/or acquire distress receptors, thereby becoming NK cell targets. Therefore, the lack of MHC class I and/or the presence of distress receptors triggers NK cell activation, resulting in cytokine production (IFN-γ) and/or cytotoxicity (via granzyme/perforin or FasL/TRAIL). Such a system allows for self-tolerance while still ensuring that deranged cells are recognized and killed. To add further complexity, NK receptors are stochastically expressed by individual NK cells. This seemingly random expression of NK cell receptors results in clones of NK cells that display enormous receptor combinations, accounting for NK cell diversity, which in turn endows these clones with the capacity to broadly recognize virally infected or malignant cells without the need for germline recombination of antigen receptors (that occurs with either B or T cells). Considering that no one receptor dominates over another, the triggering of an individual NK cell depends on the summation of the activating and inhibitory signals that a cell receives.
NK cells undergo differentiation in the lymphoid tissues and are released into the peripheral circulation. Until recently, it was assumed that once developed, NK cells are poised to kill malignant targets. However, pioneering work by Kim and Yokayama has clearly demonstrated that recognition of self-MHC (through MHC class I–recognizing NK cell receptors) is required for the acquisition of NK cell functionality.8 This process has been referred to as NK cell “licensing.” Although complex, most NK cells have a receptor that recognizes self-MHC. Therefore, a significant proportion of blood NK cells are “licensed” or cytoxic. NK cell clones that lack receptors that recognize self-MHC class I are also present within individuals and are “unlicensed.” These hypofunctional NK cells can become activated upon exposure to cytokines (IL-12, IL-15, and/or IL-18), which are present during periods of pancytopenia and infections. Once activated, these cells would not be restrained by recipient MHC (because it is lacking). In fact, in a small study by Leung et al examining autologous transplantation recipients who had NK cells expressing KIR that recognized MHC that was not present in the patient had less disease recurrence, perhaps supporting an autologous NK-mediated GVL.9 Studies are ongoing to decipher the differences between licensed and unlicensed NK cells, but such concepts are particularly relevant in the allo-HCT setting, where bacterial and viral infections are common. Therefore, there may be a dynamic interplay among recipient MHC class I, donor NK cells, and infectious complications that may modulate early, NK cell–mediated GVL reactions after allo-HCT. However, mechanisms that control these reactions have not been entirely elucidated and present opportunities to enhance GVL.
NK cells and allo-HCT
A variety of clinical studies have linked NK cells to successful outcomes after allo-HCT. For example, HCT grafts with higher numbers of NK cells are associated with a less acute GVHD (aGVHD) and chronic GVHD (cGVHD).10,11 These findings are consistent with murine studies showing that donor NK cells eradicate recipient APCs12 or alloreactive donor T cells,13 thereby preventing GVHD. Other research has linked donor NK characteristics with post-allo-HCT infectious diseases. For example, the number of KIR genes in an individual varies. Transplantation with donors that have large numbers of KIR genes was associated with fewer bacterial infections in the posttransplantation period than those with a small number of KIR genes.14 Individuals with large numbers of KIR genes typically have more activating genes, possibly implicating these receptors in the control of infections. Another emerging area involving NK cells and posttransplantation infections is the interaction between NK cells and CMV reactivation. Several groups have now demonstrated that CMV reactivation leads to the emergence of population of terminally differentiated NK cells that express NKG2C and acquire CD57 expression. These “memory” NK cells expand in response to CMV reactivation and have potent cytotoxicity and IFN-γ production in response to both acute myeloid leukemia (AML) cell lines and primary leukemic blasts.15,16 On a related note, Elmaagacli et al have shown that early CMV reactivation after allo-HCT is associated with reduced relapse rates in AML patients17 ; however, other investigators have not reproduced these findings.18,19 Therefore, the influence of CMV reaction on NK cells and GVL reactions is not yet understood.
Because NK cells make up the majority of the lymphocytes recovering early after transplantation, investigators have examined the impact of the absolute lymphocyte count or the absolute NK cell count on transplantation outcomes. Rapid lymphocyte or NK recovery (at day 28 after allo-HCT) has been linked to reductions in relapse rates or improved disease-free survival.20,21 Whether this rapid recovery represents an expansion of the mature NK cells in the graft (homeostatic expansion) or newly generated, stem cell–derived NK cells is not known and is difficult to determine. However, based on these data, methods to enhance NK cell recovery, such as the use of IL-15, may be warranted to prevent relapse and are currently being tested (“Haploidentical Donor Natural Killer Cell Infusion With IL-15 in Acute Myelogenous Leukemia [AML],” www.clinicaltrials.gov identifier NCT01385423).
Prior high-profile studies demonstrated that incompatibilities between donor KIR and recipient MHC class I (ie, KIRL mismatch) are associated with reductions in AML relapse after haploidentical transplantation,12 implying that an interruption of inhibitory KIR signals will lead to less restrained, more active NK cells. However, recent work also implicates the presence of activating KIR in the donor and protection from AML relapse. In 2 large registry reports by Cooley22 and Venstrom,23 the presence of activating KIR in the donor was associated with a significantly less AML relapse and improved disease-free survival. Although there were subtle differences between the 2 studies, both suggest that activating KIR recognize a heretofore uncharacterized ligand on AML cells. This work raises the possibility that allo-HCT donors might be selected for enhanced GVL. The feasibility of this concept is currently being testing in a clinical trial in which allo-HCT donors are prospectively being assessed for KIR gene content to determine whether, in addition to HLA typing, KIR gene content can be used in donor selection (“Selecting a Favorable KIR Donor in Unrelated HCT for AML,” www.clinicaltrials.gov identifier NCT01288222). Further evidence for a direct GVL effect of NK cells is provided by Miller et al, who demonstrated in the nontransplantation setting that lymphodepleting chemotherapy followed by the infusion of a NK cells from a haploidentical family member led to transient in vivo NK expansion and remissions in ∼30% of chemotherapy-refractory AML patients.24
Adoptive therapy with NK cells
One significant limitation is that the numbers of NK cells/kg of recipient weight obtained by leukophoresis are relatively small (∼ 2 × 107/kg). No study has linked human GVHD with NK cell infusions, so increasing the NK cell dose is one obvious approach to improving the antileukemia activity. In this regard, several promising methods are currently being tested, including expanding NK cells with artificial APCs (aAPCs) transduced with cytokines and costimulatory receptors. In particular, Imai et al have pioneered the use of K562 cells transduced with IL-15, the IL-15 receptor-α chain, and a potent costimulatory molecule, 4-1BBL. Therefore, these cells mimic the interactions that NK cells receive from aAPCs and induced significant NK cell expansion and functionality.25 Similar to this approach, work by the Lee group has used the same concept of aAPCs with the 4-1BBL costimulatory molecule, but instead of IL-15, they transduced aAPCs with IL-21/IL-21R. Before these studies, the role of IL-21 on NK cell function was controversial, but, surprisingly, these aAPCs have shown profound NK cell activation and proliferation (> 40 000-fold over 3 weeks).26 These IL-21–activated NK cells are also being tested in humans (“IL-21-Expanded NK Cells for Induction of Acute Myeloid Leukemia [AML]),” www.clinicaltrials.gov identifier NCT01787474). Still other approaches to expanding NK cells ex vivo include coculture with irradiated EBV-infected cell lines.27,28 The exact mechanism for how some EBV lines activate and expand NK cells is unknown, but this approach may be more appealing than the above studies because large numbers of stimulator cells can be created, tested, and cryopreserved for future use, perhaps leading to improved quality control and easier methods that are compliant with good manufacturing practice regulations.
Tregs: development and function
Tregs make up ∼ 5% of the peripheral blood T-cell compartment and play a critical role in peripheral tolerance. Phenotypically, they are CD4+ T cells that lack expression of the IL-7 receptor CD127 and constitutively express the high-affinity IL-2 receptor CD25 (ie, CD4+CD127−CD25+). Because activated conventional T cells (Tcons) can also express CD25, Tregs are best defined by the expression of the master regulatory gene forkhead box p3 (Foxp3). This transcription factor drives a gene expression profile that endows Tregs with a profound immunoregulatory capacity that suppresses immune activation and cell proliferation, leading to immunotolerance. Proof of the necessity of Tregs in the induction of peripheral tolerance is best evidenced by scruffy mice or in humans with IPEX (immunodysregulation polyendocrinopathy and enteropathy, X-linked) syndrome, in which the lack functional Foxp3 leads to severe and uncontrolled lymphoproliferation and autoimmunity.29 Treg ontogeny occurs through 2 distinct processes, including a thymic selection pathway resulting in so-called “natural” or nTregs, as well as the peripheral conversion of Tcons to Tregs, resulting in “induced” or iTregs (for review, see Feuerer et al30 ). The thymic pathway of Treg development involves selection steps that are similar to but distinct from the developmental events that occur during thymic selection for Tcons. Regarding the development of iTregs, recent studies show that in the presence of TGF-β and IL-2, naive T cells can be converted to iTregs, but other conditions have also been described.30
Tregs control both the antigen priming and effector stages of Tcon activation, but exactly how they mediate their immunosuppressive properties is not yet clearly defined. Tregs functionally suppress a wide range of cell types, including T cells, NK cells, B cells, and APCs, in an MHC-unrestricted manner that occurs only when the cells are in close physical proximity (contact; for review, see Shevach31 and Zimmer et al32 ). Although studies have suggested that Treg activation is required for suppression,31 more recently, studies in humans question this because resting Tregs from patients with cancer could suppress NK cell functions.33,34 Whether these observations were due to differences in the assays used, target cell types tested, or prior in vivo activation is not known.
Several potential mechanisms of Treg-mediated suppression have been put forth using in vitro T-cell assays and knockout mice, but no single pathway has been shown to be clearly implicated over another, perhaps suggesting that Tregs function through multiple mechanisms depending on the context or the cell type being suppressed (for review, see Shevach31 and Wang et al35 ). For example, Tregs may contact APCs directly, resulting in the attenuation of APC function through interactions between CTLA-4 and LAG-3 (on Tregs) binding to CD80/CD86 and MHC class II (on APCs). Alternative methods of Treg-induced suppression may be through neuropillin-1 (NRP-1), which prevents access of APCs to effector cells. Alternatively, Tregs can mount an immunological attack on several different cell types through a granzyme/perforin cytolysis pathway. Other mechanisms by which Tregs may mediate suppression are through soluble factors, such as counterregulatory cytokines (eg, TGF-β, IL-10, IL-27, IL-35) or the production of adenosine by cell-associated ectoenzymes (CD39 and CD71). Lastly, through expression of the high-affinity IL-2 receptor CD25, Tregs may act as “cytokine sinks” and effectively outcompete other cells for the available IL-2, which is essential for immune activation.
Tregs and GVHD
Preclinical murine transplantation models have convincingly established that Tregs have the capacity to prevent alloreactive T-cell responses and experimental GVHD (for review, see Riley et al36 ). Although the early data on human Tregs and allo-HCT were mixed, the majority of recent studies support a role for Tregs in the protection from both aGVHD and cGVHD.36-38 For example, stem cell grafts with a higher content of Tregs have been correlated with less aGVHD.39,40 Likewise, more rapid Treg reconstitution is associated with less aGVHD, whereas patients with delayed Treg recovery have a higher likelihood of GVHD.41,42 Matsuoka et al investigated the Tregs reconstituting early after allo-HCT, showing that the majority had an activated/memory phenotype consistent with homeostatic proliferation. Treg numbers normalized at ∼ 9 months after transplantation, but prolonged CD4 lymphocytopenia was associated with Treg apoptosis and an increased risk of cGVHD.43 Therefore, Tcons may provide IL-2 to Tregs to facilitate their survival early after allo-HCT. These observations may thereby link successful early Tcon immune recovery to reduced aGVHD via IL-2 and Tregs. Further studies from this same group demonstrated that in the setting of cGHVD, low-dose IL-2 (0.3-3 × 106 IU) could be safely administered and resulted in in vivo Treg expansion and clinical improvements in some patients.44 Considering the important role of Tregs in preventing alloreactivity, Treg-depleted donor lymphocyte infusion (DLI) has also been tested in patients with relapsed leukemia or lymphoma after allo-HCT. In a study by Maury et al, patients were eligible for Treg-depleted DLI if they did not experience aGVHD or remission after conventional, unmanipulated DLI. Of the 17 patients treated, 6 developed aGVHD after 1 or 2 doses. The development of aGVHD led to partial or complete remissions that were associated with significantly improved survival.45
Adoptive therapy with Tregs
Investigators are currently piloting a variety of different clinical grade Treg isolation techniques including magnetic bead selection and high-speed cell sorting based on surface phenotype (CD3+CD25high or CD3+CD25highCD45RA+). However, due to the relative paucity of Tregs in the peripheral blood and the high ratios of Tregs to Tcons that are potentially needed to suppress GVHD, it is anticipated that Treg expansion will be needed.35,38 Methods have been described to expand Tregs with IL-2 and either CD3 × CD28 paramagnetic beads or aAPCs that express costimulatory molecules such as CD80 or CD86 and Fc receptors (CD32 and/or CD64) that can bind antibodies such as CD3.46,47 Alternative approaches have been to generate iTregs by culturing naive T cells with TGF-β, IL-2, and rapamycin.46
Early results of the clinical adoptive transfer experiments have demonstrated safety and efficacy of Treg infusions. In the setting of double umbilical cord blood (UCB) transplantation, investigators at the University of Minnesota performed a dose escalation study using UCB-derived Tregs at doses of 1, 3, and 30 × 105 Tregs/kg at day 1 after UCB infusion. In an additional cohort, a second dose (30 × 105 Tregs/kg) was given at day 15 as well. Tregs were isolated from a partially matched third UCB unit (4-6/6 matched with the recipient) and then expanded using CD3 × CD28 magnetic beads and IL-2. In this study, Treg recipients had significantly lower rates of aGVHD compared with historical controls, whereas there was no obvious impact on either infectious complications or relapse rates.48 In an alternative approach, Di Ianni et al tested adoptive transfer of donor-derived, haploidentical Tregs which were infused during the period of chemotherapy-induced lymphocytopenia (day −4).49 The rationale was that the Tregs would expand in vivo and prevent Tcon-induced aGVHD. At day 0, patients received megadose CD34+-selected hematopoietic stem cells and Tcons at doses that would otherwise be expected to cause GVHD. This was a dose escalation study in which a fixed number of Tregs was delivered (2 × 106/kg) and the dose of Tcons was escalated (0.5 to 4 × 106/kg). Strikingly, only 2/26 patients developed significant aGVHD (≥grade 2) and there was no significant increase in relapse. Moreover, the functional recovery of T and B cells directed against pathogens was dramatically better compared with historical controls and there was no impact on the recovery of alloreactive NK cells.49
NK and Treg interactions
Studies from mice and humans clearly demonstrate bidirectional interactions between NK cells and Tregs. For example, the depletion of Tregs in mice resulted in NK cell proliferation and augmented cytotoxicity, providing evidence that Tregs suppress NK cells in vivo.34,50 Conversely, numerous human studies have shown that high numbers of Tregs within solid tumors are associated with inferior outcomes (for review, see Ghiringhelli et al33 and Pedroza-Pacheco et al34 ), perhaps suggesting an impairment in NK function. More direct proof of a Treg:NK interaction is provided by in vitro studies using coculture, where human Tregs prevented NK cytotoxicity at ratios of 1 Treg to 5 NK cells.33 In the same study, Tregs also inhibited IFN-γ production by NK cells. This suppression was maintained even with formalin-fixed Tregs and could be blocked by antibodies against membrane-bound TGF-β, strongly implicating this counterregulatory cytokine in the inhibition of NK function. Treg-induced suppression of NK cytotoxicity and IFN-γ production could be overcome by signaling through the γ-chain cytokines IL-2, IL-4, and IL-7. Mechanistically, Tregs suppress NK cytotoxicity through the reduction of NK cell–activating receptors, including NKG2D33,51 and NKp30,51 both of which play key roles in the recognition of malignant targets. Similarly, Tregs prevent IL-2–induced NKp44 expression,52 which is also used by activated NK cells to recognize malignant cells. In other studies, human plasmacytoid dendritic cells could drive NK proliferation and this was further increased by the IL-2 produced by CD4+ Tcons. The addition of Tregs did not directly impair plasmacytoid dendritic cell–driven NK cell proliferation, but did prevent Tcon-augmented NK cell proliferation, suggesting that Tregs completed for IL-2 in this system.53 Murine studies further support a role of Tregs as a “cytokine sink” of available IL-2. For example, in a diabetic model, Treg depletion led to increased NK cell proliferation and augmentation of diabetes, which, importantly, could be blocked by IL-2–specific antibodies.54 Complementary studies by Gasteiger et al showed that Tregs prevented the IL-2–dependent NK maturation and activation in a tumor and viral infection model.55 Moreover, upon depletion of Tregs, NK cells were more responsive to targets that were missing self-MHC, a situation analogous to KIR ligand mismatch in humans.56 Still further proof of this, our own preliminary studies showed that Tregs can impair NK cell function when IL-2 is present in the culture media, but not when IL-15 is present (V. Bachanova, M.R.V., J. S. Miller, unpublished data, 2013).
Although the majority of data support a Treg-mediated suppression of NK cells, there are some data to suggest NK:Treg cross-talk is bidirectional and that NK cells can impair Treg expansion and function. For example, in a tuberculosis model, NK cells cultured with mycobacterium-exposed APCs impaired Treg proliferation and could even kill expanded Tregs in an NKG2D-dependent manner.57 These results suggest that under steady-state conditions, Tregs negatively modulate NK cells, but during infectious challenges, the opposite occurs, perhaps as a method to respond to such challenges. Other studies have documented that NK-derived IFN-γ can prevent the expression of Foxp3 and the peripheral conversion of Tcons to Tregs.58
Potential opportunities
Tregs play an important role in allo-HCT by taming alloreactivity and quelling GVHD, but they also may impair NK (or T-cell)–mediated GVL. Although the early human studies of Treg adoptive transfer do not necessarily support this claim,48,49 the numbers of treated patients are small and the diseases heterogeneous. Moreover, the dose of Tregs infused is relatively small, leaving the impact of Tregs on NK cell function and relapse an open question. Understanding the interactions between Tregs and NK cells presents possible opportunities to modulate either GVL or GVHD. Considering that Treg depletion in the setting of DLI improved GVL responses45 and a similar approach (of Treg depletion) could be used to modify hematopoietic stem cell grafts. Other strategies to augment GVL responses may involve transient Treg eradication using CD25-directed antibodies (diclizamab) or immunotoxin-conjugated IL-2 (denileukin diftitox). In fact, our recent studies suggest that that denileukin diftitox before haploidentical NK cell infusions led to NK cell expansion in the blood of individuals 14 days after infusion, a marker of disease response (V. Bachanova, M.R.V., J. S. Miller, unpublished data, 2013). Still other approaches to augment GVL reactions after allo-HCT may to use drugs that selectively impair Tregs. Recently, lenalidomide and pomalidomide have been shown to impair Treg expansion and function in vitro. Although the mechanism of the suppression is not entirely clear, in vitro studies show that these immunomodulatory drugs impaired Foxp3 expression but not TGF-β production.59 Lenalidomide has recently been tested in the posttransplantation period in a group of myeloma patients undergoing allo-HCT. Briefly, lenalidomide was started at day 100-180 after transplantation in this dose escalation study. The maximum tolerated dose of lenalidomide was 5 mg/d × 21 days for an anticipated 4 cycles. aGVHD occurred in 38% of patients, mostly during the first 2 cycles. Some patients showed changes in the immunological parameters, including NK and Tcon activation and a reduction in Tregs. Patients who showed these changes had superior survival.60
In summary, donor NK cells have the capacity to recognize and kill leukemia cells. The rapid recovery of NK cells early after allo-HCT is implicated in GVL responses. Conversely, Tregs restrain immune responses and mediate tolerance. Murine studies and human in vitro experiments support the assertion that Tregs antagonize NK responses. Whether this occurs in humans after adoptive transfer or allo-HCT is an open question. Although some studies support an augmented NK cell response in the absence of Tregs, the clinical trials are just now being performed. Regardless, manipulation of these 2 cell types using the methods described have the potential to not only reduce aGVHD, but also to augment GVL. We look forward to clinical studies aimed at accomplishing this task.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Michael R. Verneris, MD, University of Minnesota, MMC 366, 420 Delaware Street SE, Minneapolis, MN 55455; Phone: 612-626-2961; Fax: 612-626-3941; e-mail: verneris@umn.edu.
