
Abstract
The application of high-throughput genomic approaches in lymphomas has generated a wealth of data regarding the molecular underpinnings of these cancers. In this review, key findings from recent studies are discussed, as well as the genetic heterogeneity that underlies common lymphomas including diffuse large B-cell lymphoma, Burkitt lymphoma, and chronic lymphocytic leukemia, and the implications for identifying new therapeutic opportunities and personalized medicine.
Introduction
Lymphomas represent a diverse group of malignancies comprising at least 48 distinct types.1 The classification schemes for lymphomas continue to evolve as we learn more about the molecular underpinnings of these tumors and their presumed cells of origin. However, even within an individual diagnosis, there is usually considerable heterogeneity2 with respect to clinical outcome, genetic alterations, and the expression of commonly assayed markers. Therefore, discerning the correct diagnosis and prognosis for an individual patient with lymphoma remains a daunting clinical challenge.
The advent of high-throughput genomics technologies has provided new opportunities for understanding the molecular building blocks of cancers. The 2 main tools of these technologies, microarrays and high-throughput sequencing, have led to a sea change in our approach to tumor-based measurements. Rather than individual measurements of gene expression or genetic alteration, it is now possible to simultaneously assay these in a genome-wide fashion using genomics technologies. The application of these powerful technologies has yielded several insights into the molecular processes underlying lymphomas.
Although it is not practical to describe the findings in every lymphoma type in sufficient detail, we describe the application of genomics in 3 separate lymphoid tumors, diffuse large B-cell lymphoma (DLBCL), Burkitt lymphoma (BL), and chronic lymphocytic leukemia (CLL), to highlight these approaches and discuss the opportunities and challenges that lie ahead.
DLBCL
DLBCL is the most common form of lymphoma in adults, accounting for approximately 25 000 new cases and nearly 10 000 deaths per year in the United States alone.3 Although more than half of the patients are cured4 with current chemotherapy regimens, the majority of the patients who fail to respond will succumb to the disease. It has proved difficult to develop new therapies for patients with DLBCL. An important reason for the failure of many clinical trials in DLBCL may be the approach to the disease as a single entity, even though it is known to be molecularly and clinically heterogeneous.
The most widely adopted clinical risk stratification of DLBCL remains the International Prognostic Index (IPI),5 a multivariate score composed of age, stage, performance status, serum LDH, and extranodal disease sites. The IPI score remains the accepted standard for clinical stratification nearly 2 decades after it was proposed, being used in several clinical trials and offering a simple method for risk stratification of patients with the worst prognosis. The shortcoming of this scoring system is that the biology underlying the prognosis remains obscure.
Molecular approaches using gene expression profiling to subgroup DLBCLs have added considerably to our understanding of the heterogeneity of the disease. For example, several gene expression profiling studies of patients with DLBCL demonstrated that the tumors comprised at least 2 distinct diseases with different cells of origin, distinct cytogenetic differences, and different response rates to anthracycline-based chemotherapy regimens. One subgroup, termed germinal center B-cell-like (GCB) DLBCL, shares characteristics of normal germinal center B cells, including the expression of genes such as BCL6 and CD10. The other subgroup, termed activated B-cell-like (ABC) DLBCL, expresses genes associated with B-cell activation, including Pim-1 kinase and IRF4. Five-year survival rates for patients with ABC and GCB are significantly different, with a rate of nearly 75% for GCB patients but less than 30% for those with ABC DLBCL.6
Other approaches to subgrouping DLBCL using gene expression profiling have also been proposed. In particular, another scheme uses gene expression profiling to identify 3 distinct subgroups of DLBCL including those related to B-cell receptor (BCR) activation, host response, and oxidative phosphorylation.7 Although these subgroups did not have prognostic relevance at the time they were described, the advent of BCR-targeted therapies may provide relevance for these findings.8
Additional gene expression signatures that reflect distinct disease processes have been also described.6,7,9,10 These processes, including those related to proliferation, are also independently associated with survival. However, the biological mechanisms and genomic alterations that are responsible for these changes in gene expression are still poorly understood. In addition, most gene expression signatures have been derived from single studies using microarray-based gene expression. Evaluating and extending these signatures in RNA-sequencing-based gene expression measurements could illuminate additional signatures by including noncoding RNAs11 and defining additional subtypes that are biologically relevant.
Parallel efforts have attempted to integrate outcome associated gene expression into clinically useful prognostic panels with some success. In particular, an assay based upon quantitative PCR of 6 genes (LMO2, BCL6, FN1, CCND2, SCYA3, and BCL2)12 that combines aspects of the prognostic molecular subgroups and outcome-associated gene expression signatures has been applied in the clinical setting.
More recently, high-throughput sequencing has defined the genetic landscape of mutations in DLBCL.13-15 These efforts have revealed that several hundred gene mutations underlie the disease. Even the most common mutations in DLBCL (eg, MLL2 and MLL3) affect only a minority of patients. These findings highlight the difficulties in developing newer therapies for this disease and suggest that targeted approaches will rely on a better understanding of the molecular makeup of each individual patient's tumor.
BL
BL is a highly aggressive form of non-Hodgkin lymphoma that is characterized by deregulation of the MYC (c-myc) gene. Although a relatively uncommon disease, with an annual incidence of ∼ 2000 in the United States,3 it is nevertheless important not only because of its toll on patients, but also because it is a major disease model for molecular studies in many other types of lymphoma and other cancers. The role of MYC in malignancies was first discovered in BL.16 BL tumors and cell lines continue to be used for the exploration of oncogenes and the biology of normal and malignant B cells.17,18
The diagnostic distinction of BL from DLBCL can be difficult because of overlapping morphology, immunophenotype, and cytogenetics.19 The difficulty and importance of obtaining the correct diagnosis in BL was highlighted by the experience of the Cancer and Leukemia Group B trial CALGB 9251,20 in which nearly half of the 100 patients with an assigned diagnosis of BL were found to have another diagnosis upon careful pathology review. However, the distinction is clinically important because of major differences in the treatment of the 2 diseases. BL requires the use of intensive chemotherapy regimens that carry a higher risk of morbidity and mortality and such treatments are not usually used in DLBCL.
The application of gene expression profiling in BL has shown that BL tumors have a distinct molecular phenotype that allows their distinction from other aggressive lymphomas, including DLBCL.19,21 These data indicate that a group of genes associated with MYC target genes, germinal center differentiation, NF-κB expression, and MHC class-I expression identifies BL with high certainty.
More recent work has identified the genetic makeup of BLs through the application of high-throughput sequencing.22-24 This work has enabled the identification of ID3 as a key tumor suppressor gene that appears to serve as a negative regulator of MYC-driven proliferation. More than 70 other genes were recurrently mutated in the disease, including TP53, PTEN, GNA13, and EZH2, suggesting that multiple genetic pathways can culminate in the observed BL phenotypes.
The relative uncommonness of BL presents a real challenge in designing and accruing appropriate clinical trials. Therefore, approaches that identify the molecular features of these tumors will be needed to better match these patients with therapeutic opportunities and/or clinical trials, particularly in the relapsed setting.
CLL
CLL is the most common form of adult leukemia, affecting > 8000 new patients each year. Although the course of the disease can be indolent, a large proportion of patients with CLL will succumb to their disease. In addition to Rai/Binet staging,25,26 which use clinical variables measuring progression, the mutation status of the IGH gene,27 CD38 expression,27-29 and ZAP70 expression30-32 have been shown to be associated with prognosis.
Cytogenetic abnormalities have been extensively tested in CLL, with 17p and 11q deletions conferring the worst prognosis, and trisomy 12, normal karyotype and 13q deletion conferring a relatively favorable prognosis.33,34 The application of high-throughput sequencing in CLL35,36 has identified several recurrently mutated genes in CLL, including those related to DNA-damage response and cell cycle progression (eg, TP53 and ATM), RNA splicing (eg, SF3B1 and DDX3X), and Notch signaling (eg, NOTCH1). Mutations in several of these genes, including TP53,37 NOTCH1,38 and SF3B1,39 are associated with poorer prognosis. However, the role of most genetic mutations and prognosis in CLL remains to be defined.
Intriguingly, it is known that nearly all CLLs arise from monoclonal B-cell lymphocytosis (MBL),40 a surprisingly common condition that rises in incidence with age. Although the majority of patients with MBL will experience a benign course, the genetic risk factors underlying the progression of MBL to CLL remain largely unknown. If the genetic mutations associated with progression to CLL can be defined, it would offer the possibility for risk-adapted methods for surveillance of MBL and potential intervention at an early stage of the disease.
Heterogeneity of lymphomas demands a new paradigm for clinical translation
The common thread that unites the application of genomic technologies in virtually every cancer, including lymphomas, is the uncovering of a marked genetic heterogeneity within a single diagnosis. This heterogeneity presents a challenge and a potential opportunity for developing new therapeutics in the disease. Lymphomas themselves are diverse with regard to heterogeneity. Some tumors, such as Waldenstrom macroglobulinemia41 and hairy cell leukemia,42 have common mutations that affect the majority of the patients, suggesting immediate therapeutic targets in these entities. However, even in these entities, the additional mutations that cooccur in the tumor can be very different from patient to patient. In most other lymphomas, the heterogeneity of the disease suggests that no matter the targeted agent being used in a particular lymphoma type, only a small proportion (typically < 25%) of the individuals are likely to harbor a mutation relevant to the targeted pathway.
This observed heterogeneity also offers new opportunities to identify those patients who are most likely to respond to targeted therapy. For example, this approach has been applied successfully in the treatment of melanoma with the BRAF inhibitor vemurafenib43 and in colorectal cancer, in which patients with KRAS mutations are unlikely to respond to cetuximab.44 Rare mutations in IDH1,45 BRAF, and KIT are also observed in lymphomas, providing new potential therapeutic opportunities for patients with those mutations.
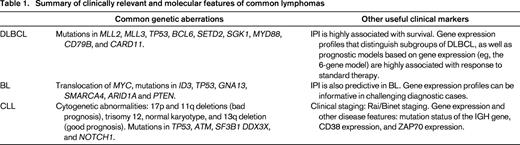
As lymphomas are classified and subclassified extensively based on their molecular features (Table 1), there has been a corresponding rise in the identification of intermediate entities that harbor the defining features of more than one lymphoma type. Examples of such entities include those gray zone lymphomas that are intermediate between Hodgkin lymphoma and DLBCL, as well as those between BL and DLBCL. The appropriate diagnosis and treatment of these entities remains a topic of widespread disagreement. The comprehensive profiling of lymphomas will likely increase the number of cases that cannot be classified with great certainty. For example, the gene expression profiling-based distinction of DLBCLs created an additional category of unclassified DLCBLs that did not previously exist. Patients with tumors belonging to these unclassified entities might benefit from having their tumors profiled and from participating in a tumor registry.
Conclusion
The application of high-throughput technologies has created new opportunities and challenges in the risk stratification and treatment of patients with lymphomas. Ultimately, there is a simple yardstick to measure progress in this rapidly expanding field: “Does it lead to a better outcome for the patient?” The evolving story of heterogeneity in the molecular makeup of lymphomas will demand a variety of risk-adaptive strategies for clinical trial design and assessment of response. Patients likely to respond to the inhibition of PI3 kinase inhibitors46 could be distinct from those likely to respond to BRAF inhibitors and other targeted therapies. In the short term, the application of genomics has complicated our understanding of lymphomas and other cancers by identifying a plethora of genetic aberrations that characterize these tumors. In the long term, these studies form the basis of personalized medicine in which the genomic profile of each patient's tumor might be used to guide therapy that is most likely to benefit that patient.
Disclosures
Conflict-of-interest disclosure: The author declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Sandeep S. Dave, MD, Department of Medicine, Duke University School of Medicine, DUMC 3382, Durham, NC 27710; Phone: 919-681-1922; Fax: 919-684-4777; e-mail: ssd9@duke.edu.
