Abstract
New epidemiological findings recast pain in sickle-cell disease (SCD) as being more often a chronic manifestation than was previously thought, although acute pain is still the hallmark of the disease. SCD pain intensity, the number of painful locations, and the frequency of hospitalizations due to SCD pain may worsen with age. In adults and even in children, the quantity and severity of SCD pain may be vastly underestimated, because most of the “iceberg” of SCD pain is “submerged” at home, and only the tip of the iceberg is seen by health care providers when acute SCD care is rendered in emergency rooms and hospitals. Implications of this “iceberg phenomenon” are significant for clinicians, researchers, employers, policy makers, and the public. Nevertheless, both emergency room and hospital utilization for SCD pain remain prevalent. Often, utilization recurs early, perhaps emblematic of poor acute pain management. New data show the protean impacts of SCD pain on health-related quality of life, sleep, and psychological and social health. The relationship of the severity of SCD pain to the severity of underlying sickle vasculopathy is unclear, but epidemiologic evidence and patient descriptors suggest a temporal evolution of pain mechanisms. At first, increasingly worse nociceptive pain from vaso-occlusion and local lesions may evolve over the first two decades of life. Then, in the third and following decades, central neuropathic pain may also evolve due to past and continuing nociceptive stimuli. New findings confirm environmental contributors to SCD pain, including seasonal (colder) temperatures, barometric pressure, and wind speed.
Epidemiology of SCD Pain
Frequency and Chronology of Pain
Pain is the most common symptom reported by the approximately 100,0001 Americans with sickle-cell disease (SCD), and this pain may be disabling. Previously, SCD hospitalizations and medical contacts were described in longitudinal studies. “Pain” as defined in these studies was found to be infrequent2 and to be the result of vaso-occlusive crises: severe, acute pain from vaso-occlusion and its inflammatory and ischemic consequences.
However, SCD pain may actually occur quite frequently. Adult respondents in the Pain in Sickle Cell Epidemiology Study (PiSCES) reported SCD pain on 54.5% of the 31,017 days surveyed. Importantly, 29.3% of respondents had pain on greater than 95% of the days surveyed.3 Dampier et al. studied children and adolescents (ages 6–21 years) with SCD for a total of 18,377 days, and found less frequent, but still surprisingly common pain; those studied reported 514 distinct SCD pain episodes occurring over 2592 days and 2326 nights.4 In a related report, children and adolescents reported SCD pain alone 8.4% of the time (1515 d), whereas other pain occurred 2.7% of the time (490 d), and both SCD and other pain 5.7% of the time (1041 d).5 These results show that acute pain is definitely the hallmark of SCD, but that chronic pain occurs far more often than was previously realized.
Severity of Pain
Pain in SCD is not only common, but also severe. In adults enrolled in PiSCES, SCD pain intensity was measured on a 0 to 9 numeric scale, but was also classified into three mutually exclusive severity categories: pain resulting in emergency room or hospital utilization (mean intensity, 5.9 ± 0.1); pain described by the patients as a crisis, but not resulting in emergency room or hospital utilization (mean intensity, 5.0 ± 0.1); and pain not described as a crisis (mean intensity, 3.9 ± 0.1).
Patients who described more frequent pain also described more intense pain on average. Those who described being in pain on 96% to 100% of days reported a mean pain intensity of 5.1 ± 0.2 on pain days and 6.2 ± 0.2 on crisis days, whereas those who described pain on 5% or fewer of days reported an intensity of 3.5 ± 0.4 on pain days and 4.5 ± 0.6 on crisis days. Opioid use was strongly correlated with pain intensity.
In the above childhood study, analgesic medication was taken on 88% of the reported pain days and 76% of reported pain nights. A single oral analgesic was used on 58% of these days. On the remaining days, multiple analgesics were used in a variety of combinations. More frequent analgesic dosing was reported on days with more intense pain. In a related report on this sample, pain intensity was reported on a 0 to 10 scale, and was 3.9 ± 0.5 in 12 younger children (ages 5–9 years), but 5.3 ± 0.4 in 18 older children (ages 10–19 years).6
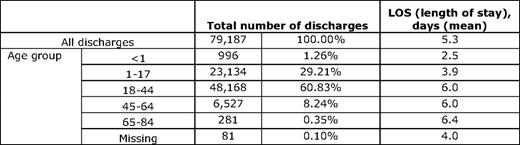
Researchers have organized evidence supporting a theory of distinct phases (e.g., evolving, inflammatory, resolving) of a painful vaso-occlusive episode that results in hospitalization, and the underlying biology associated with rises and falls in pain severity during such an episode.7 Recent evidence adds to this theory. Even after hospital discharge, resolution of a painful episode may take quite some time, and may be associated with significant dysfunction, resulting in more days taken off of school or work than necessitated by hospitalization.8 Further, readmission soon after hospitalization is not uncommon.9 The above theory predicts that episodes typically last for 7 to 9 d, but Table 1 shows that, nationally, hospital lengths of stay are only 5 d when averaged overall, worsening from 3 to 6 d with age. Thus, readmission could represent failure of an original episode to resolve, inadequate supply of analgesia for pain occurring after discharge, or even hyperalgesia from opioid withdrawal.
Location(s) of Pain
In adults enrolled in PiSCES, an average of 3.3 out of 16 sites (25%) were painful on the days surveyed. The number of pain sites varied by age, depression, frequency of pain days, crisis, and unplanned hospital/emergency room utilization. Pain was located in the lower back, knee/shin, and hip on average for more than one-third of pain days, while jaw and pelvis pain were infrequent (fewer than 10% of days). The odds of patients self-designating pain as a crisis were greater (odds ratio [OR] > 1.5) when pain was in the arm, shoulder, upper back, sternum, clavicle, chest, or pelvis. Unplanned utilization was substantially more likely (OR > 2.0) when pain was in the sternum, clavicle, or chest.11 Dampier et al. found fewer pain sites on average in children: 2.2 ± 0.3 in younger children and 1.9 ± 0.2 in older children.
Responses to Pain
Visiting a health care provider for pain relief is a rational response. Not surprisingly, pain is responsible for the majority of SCD medical visits. Platt et al. studied rates of medical visits for pain among 3578 patients in the Cooperative Study of Sickle Cell Disease (CSSCD). Above age 20, higher-utilizing adults were at higher risk of death. Utilization due to SCD pain increased as patients grew older, from 0 to 30 years, and declined thereafter.2 Combining the above results with studies showing that the frequency, severity, and number of sites of pain are greater in adults than in children suggests that the frequency of pain rises with the androgen-associated rise (especially in males) of hematocrit with the onset of puberty.12
Despite the above statistics, the utilization of formal health care resources in response to SCD pain is infrequent when the denominator of all pain experienced by patients is included. Most SCD pain, even “crisis” pain, is managed at home, without emergency room or hospital utilization.13 Further, SCD pain that does result in health care utilization may be indistinguishable from pain managed at home. In PiSCES, while pain (with or without crisis or utilization) was reported on a total of 54.5% of days, crises without utilization were reported on 12.7% of days, and utilization for pain on only 3.5%. Significantly, the rate at which utilization days coincided with crisis days was far below what would be the expected if the two were essentially identical.3
A substantial minority of SCD patients are high utilizers of hospitals and emergency rooms, and in fact are responsible for most of the visits; these patients are more severely ill, as measured by laboratory variables, pain, distress, and/or quality of life.14,15 In PiSCES, after controlling for severity and frequency of pain, high emergency room utilizers (35% of the sample) did not use opioids more frequently than other SCD patients.15
A burst of literature has appeared that has further characterized the utilization of hospitals and emergency rooms for SCD pain. Most studies used national, administrative claims-based datasets containing unselected, highly representative patient samples and/or surveys.16 For example, a study of administrative claims data from eight geographically dispersed states representing 33% of the US population with SCD (21,112 patients) updates Platt's original, smaller CSCCD report of decades ago. The CSSCD relied on recruited patients and used primary data collection to measure utilization events.2 The 8-state study reported a rate of acute care encounters, 2.59 per patient per year, that was 2 to 3 times higher than in the CSCCD. There were 1.52 hospitalizations and 1.08 treat-and-release emergency room visits per year. Approximately 29% of the population had no encounters, while 16.9% had 3 or more encounters per year. In the CSSCD, 40% of the population had no encounters in a year, while 5% accounted for a large proportion of utilization. Further, re-hospitalization rates for the 8-state study were frequent: 22.1% and 33.4% at 14 and 30 d, respectively. Both utilization rates and re-hospitalization rates were highest for 18- to 30-year-olds.17
Other studies have shown that hospitalizations for SCD pain are more frequent and hospital lengths of stay are longer among older children than younger children,18,19 although the percentage of all emergency room visits resulting in hospitalization actually dropped in one study among older compared with younger children.20 SCD children also demonstrate unmet health care access needs and show higher odds of frequent severe headaches or migraines, intellectual disabilities, regular use of prescription medication, and fair or poor health status compared with African-American controls.21
Another rational response to pain is analgesic use, and in SCD, use appears rational. In PiSCES, home opioid analgesic use varied widely: a total of 39 out of 232 (16.8%) of patients studied did not use opioids at home during any of the days surveyed, whereas 88 (37.9%) used them on 1% to 49% of days, and 104 (44.8%) used them on 50% or more of days. Opioid use independently predicted pain, crises, and hospital and emergency room utilization.3
In children, Dampier et al. found that cognitive-behavioral or physical pain-management activities were used alone on 7.5% of pain days and with analgesics on 77%. Female gender and increasing pain intensity were associated with an increased number of pain-management activities used. Increasing pain intensity was also associated with the use of several specific pain-management activities.22
Impact of SCD Pain
Depression, Psychological, and Neurological Impacts
Many studies have documented that depression and anxiety disorders are common in SCD and may be correlated with SCD symptoms.23 More recent, however, is the notion that chronic pain, via neurological changes in the prefrontal cortex (see discussion of neuroplasticity and remodeling below), may affect emotion. This could as easily be true in SCD as in any other chronic painful disease. For example, having chronic back pain or fibromyalgia is also associated with decreased grey matter in the prefrontal cortex, which may be associated with a decreased ability to inhibit the experience of pain.24
Health-Related Quality of Life
New research reconfirms that individuals with SCD have poor baseline health-related quality of life, and painful episodes have a further negative impact.25 This is most striking in the physical functioning and pain domains. Worse health-related quality of life in SCD patients is associated with disease severity and pain intensity.26
Sleep
Aside from emerging relationships between nocturnal hypoxia, sleep-disordered breathing (including sleep apnea), and pulmonary hypertension identified in SCD patients,27 anecdotal reports from patients and new research suggests that there are sleep disturbances related primarily to SCD pain, and vice versa, in SCD patients.28
Etiology of SCD Pain
Local Mechanisms
The above epidemiologic research, combined with new research in other disease states, suggest the need to revisit theories about how SCD pain evolves over time and to revisit approaches to pain management in SCD. The most important finding is that pain in SCD is more often chronic than was previously realized, although acute pain is still the hallmark SCD presentation. Figure 1 is a conceptual, hypothetical model of pain in SCD, beginning with age 120 months (10 years) and showing evolution with time. The first two types of pain, as depicted by the legend to Figure 1, represent known, recurrent, nociceptive pain (defined below) experienced due to vasculopathic and necrotic conditions, respectively. The final two types of pain described in the legend depict neurological pain manifestations hypothesized to develop later in life in response to prior recurrent nociceptive pain or in response to opioid therapy.

Conceptual model of etiologies and temporal evolution of pain in SCD. Multiple etiologies may be present in any one patient.
Conceptual model of etiologies and temporal evolution of pain in SCD. Multiple etiologies may be present in any one patient.
Undoubtedly, the ultimate etiologic source of pain in SCD early in life is vaso-occlusion, which is ubiquitous. Vaso-occlusion is represented by the solid line in Figure 1. However, SCD pain episode rates vary between subjects, even within genotype. Further, the location, timing, and severity of pain episodes may be unpredictable. Logically, most attribute this variability to underlying variations in vaso-occlusion and their consequences. Epidemiologically, pain is paroxysmally severe, resulting in emergency room and hospital utilization, but it is also often prevalent at an intensity that is managed at home. It follows that vaso-occlusion may be chronic and may always be present at some low level. For some patients, the pain represented by the line in Figure 1 may never go to an intensity of zero.
Vaso-occlusion results in a complex progressive cascade of tissue ischemia and inflammation. This appears to result from impaired rheology due to the sickled shape of polymerized red blood cells in SCD, as opposed to their otherwise flexible, biconcave morphology that makes them capable of reaching arterioles and capillary beds of muscle and organ tissue. A conformational change in hemoglobin S also impairs its oxygen-carrying capacity compared with normal hemoglobin A. A panoply of processes result, including red-cell sickling, inflammation and adhesion, coagulation activation, stasis, deficient bioavailability and excessive consumption of nitric oxide, excessive oxidation, and reperfusion injury. Together, these processes are called sickle-cell vasculopathy.29 Pulmonary hypertension, priapism, fetal wastage and growth retardation, autosplenectomy, and chronic renal disease are all well-documented in SCD. Vasculopathy contributes to tissue ischemia and necrosis in these and other complications of SCD, potentially ending in organ failure of the spleen, kidneys, heart, lungs, brain, liver, and bone, and in early death.30
Reliable biomarkers of chronic vasculopathy in SCD are being researched31 ; however, biomarkers able to distinguish the severity and temporal changes in stimuli underlying pain exacerbations have still not been identified. Dampier et al.'s longitudinal studies of SCD children demonstrate similar clinical features and similar predictive hematologic parameters of home-managed versus acute care-managed pain, implying a single underlying pain stimulus of variable intensity.6 However, no studies show body-location-specific changes in biomarkers associated temporally with changes in pain at that location. To date, we cannot “predict a crisis” reliably using biomarkers.
At least early in life, the neurologic mechanism linking underlying pain stimuli to pain experience in SCD is that of nociception. Nociception requires that pain be due to an identifiable pain stimulus that is potentially or actually damaging. The nociceptive nervous system involves the peripheral nerves, spinal cord, brain stem, thalamus, and cerebral cortex, and links recognition of a damaging stimulus to an adaptive behavioral response.32 A noxious or potentially damaging stimulus is linked to a sensation that is perceived as intense and unpleasant (pain), which is designed to allow an association of this sensation with something that must be avoided.
In acute pain, hyperalgesia ensures that contact with or movement of the affected structure will be minimized until healing is complete. This is a critically important adaptive mechanism in SCD, where widespread tissue damage is common. It involves four major pathophysiologic processes: transduction, transmission, modulation, and perception.33 This innate physiological response is likely repeated countless times during vaso-occlusive episodes in the first two decades of life. This repetitive stimulus is suspected of being one of the key precursors to the chronic pain experienced by some SCD patients later in life.
A second source of nociceptive pain in some SCD patients comes from a clearly localized source: tissue necrosis. Bone infarction of the spine and long bones, as well as avascular necrosis of the hip and shoulder, are frequent in SCD, and are localizable by clear radiographic findings. Bone infarction may wax and wane, and may be physically manifested by heat, swelling, rubor, tenderness of the extremities, or dactylitis. Chronic leg ulcers in SCD are painful. When they appear, they generally are in locations consistent with venous stasis rather than arterial insufficiency, and they anecdotally may sometimes respond to elevation, which is consistent with venous stasis ulcers. Pain from splenic infarction is localizable and discrete, and is often acute and disabling. The stimuli producing this nociceptive pain may be chronic and ongoing or may wax and wane.
Neuropathic Pain Mechanisms and Neuroplasticity
Chronic pain has been defined as that resulting when there is an uncoupling of nociceptive processes from noxious stimulation or healing.32 Chronic pain has also been defined temporally as pain lasting for more than 6 months.34 Figure 1 shows that, by either definition, pain in SCD may be chronic. In addition to nociceptive pain, chronic neuropathic pain may well complicate acute pain in SCD. In general, neuropathic pain occurs as a result of malfunction or injury in the peripheral or central nervous system. Temporally, it may be acute or chronic. Neuropathic pain has been demonstrated in many diseases, notably herpetic neuralgia,32 diabetic neuropathy,35 fibromyalgia,36 chronic low back pain,37 phantom limb pain,38 and functional bowel disease.39 In SCD, neuropathic pain may result from prior nociceptive painful episodes. Specifically, in other disorders, nociceptive input can sensitize supraspinal areas involved in pain processing, causing them to become more active during future events of noxious stimulation, which is expressed phenotypically as a lowering of the pain threshold.24 Subsequently, there may actually be parallel anatomical brain changes in the surrounding glia (astrocytes and microglia) called neuroplasticity or remodeling, which contribute to the maintenance of an altered, neuronal phenotype. While Figure 1 emphasizes this mechanism as producing chronic pain, it may be that the severity of acute nociceptive pain episodes is intensified by this same neuropathic mechanism after remodeling has occurred.
Neuropathic pain can be inferred when patients use certain pain descriptors (e.g., burning, tingling, shooting, numbness, and lancinating) to describe their pain.40 It can also be documented experimentally by demonstrating sensations of pain in the absence of an experimental stimulus and/or by demonstrating abnormal (hyperalgesic) pain responses from standardized pain stimuli.41 Using these criteria, there are few studies so far demonstrating neuropathic pain in SCD. Epidemiological studies of pain have not asked patients to use pain descriptors, nor have they included psychophysical testing. One study of transplanted patients ages 16 to 45 years with severe SCD who achieved cure showed that they still had pain after transplantation, which lasted for months.42 This pain was attributed to opioid withdrawal, but testing was not done to confirm if the pain was possibly neuropathic. A recently published phenotypic classification system for SCD allowed for the existence of neuropathic pain due to “nerve ischemia from vaso-occlusion,”43 but listed only one publication reporting neuropathic pain in SCD, and it referred only to peripheral neuropathy.44 Only one recent study has employed pain descriptors, which revealed that SCD patients choose descriptors commonly used to describe neuropathic pain and nociceptive pain to describe their SCD pain.45
Figure 1 shows that neuropathic pain in SCD likely does not manifest itself until early or late adulthood, and only in patients with sufficient prior nociceptive pain; longitudinal studies that begin in childhood may be necessary to confirm these hypotheses. If they are confirmed, then further studies would be required to determine if more effective disease and pain management in adolescents and young adults reduces the incidence and severity of both acute and chronic SCD pain in older adults.
Finally, Figure 1 shows a fourth potential source of SCD pain: that from chronic and/or acute opioids given to treat pain. Opioid-induced hyperalgesia is considered to be due to the administration of opioids, and is different from the hyperalgesia described above, which is provoked by prior nociceptive stimuli. Opioids temporarily reduce central responsivity to any pain stimulus. Although not yet proven in SCD, evidence from experimental laboratory studies and opioid treatment in other diseases suggest that acute or chronic opioid administration may cause hyperalgesia.46 The SCD pain stimulus no doubt continues in the face of palliation of pain using opioids. In cancer, this effect is usually short-lived, ending in the death of the patient, so any opioid-generated hyperalgesia is less important; however, in SCD, opioid-induced hyperalgesia could go on for decades, depending on the severity of the pain and the chronicity and intensity of opioid therapy.33 Like neuropathic pain from prior nociceptive pain, opioid administration in SCD may alter future nociceptive pain, exaggerating the innate hyperalgesic response that is part of nociception.
Environment: Weather, Barometric Pressure, and Wind Speed
Two multi-site studies have continued research into the effects of climate and environment on SCD pain. The first failed to find a significant relationship between temperature and the occurrence of painful episodes, but did find that higher wind speeds during the preceding 24 h were associated with the onset of pain.47 Conversely, the Multicenter Study of Hydroxyurea found positive, direct associations between other climate conditions and SCD pain. Adults with SCD reported exacerbated SCD pain during seasonably colder temperatures, but not during days with lower barometric pressure. Colder seasons were significantly associated with greater pain intensity but not frequency. Higher average monthly temperatures were statistically significantly associated with lower pain intensity and lower pain frequency. Both pain intensity and pain frequency were statistically significantly higher when the average monthly barometric pressure was higher. Northern geographic residence was not statistically associated with higher pain intensity and frequency compared with southern residency, although the differences did suggest a trend.48
Genetic Predictors
Beyond the scope of this article is a discussion of genetic predictors of response to SCD therapies.49 However, recent data provide evidence that pain phenotypes in SCD have more complex underlying genetic explanations than simple hemoglobin type. Within the hemoglobin S genotype, haplotypes of the S gene appear to be related to specific phenotypes of the disease, including pain phenotypes.50
Psychological Predictors
In addition to being an outcome of pain in SCD, psychological factors may also be predictive of pain. Cognitive factors (e.g., catastrophizing, or irrational thought that something is far worse than it actually is) and psychological factors (e.g., depression and coping) influence the central representation of pain.51 We found that SCD subjects had higher mean catastrophizing scores than those found in studies of other chronic pain conditions that are not lifelong or life-threatening.52
Two recent studies advanced the understanding of the impact of depression on pain.53,54 The latter study found that depressed subjects had pain on significantly more days than non-depressed subjects. When in pain on non-crisis days, depressed subjects had higher mean pain, higher distress from pain, and more interference from pain.52
Over two decades ago, coping strategies among SCD adults were found to be important predictors of pain and adjustment. Individuals with high scores on the negative thinking and passive adherence categories had more severe pain, were less active and more distressed, and used more health care services. Individuals with high scores on coping attempts were more active during painful episodes.55 A more recent study has found that active coping is positively associated with pain frequency, while passive adherence coping is related to pain intensity. Psychological coping was shown to be unrelated to health service utilization. The degree of impairment was associated with affective coping.56
Summary
Taken together, the above research advances lead to the conclusion that pain in SCD is far more common and severe, with more significant effects on all aspects of life, than was previously reported. For many SCD patients, pain is the rule rather than the exception, and is likely multifactorial in etiology, not simply vaso-occlusive. Pain prevalence is probably vastly underestimated by health care providers, resulting in misclassification, distorted communication, undertreatment, possible unnecessary loss of employment and productivity by patients and caregivers, and mistrust even from family members and friends of patients. While a discussion of the measurement and treatment of SCD pain is beyond the scope of this article, results from the above epidemiology and etiology studies demand that clinicians, researchers, employers, policy makers, and the public rethink the measurement and treatment approaches to pain in SCD.
Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests. Off-label drug use: None disclosed.
Correspondence
Wally R. Smith, MD, Professor, Division of General Internal Medicine, Virginia Commonwealth University, P.O. Box 980306, Richmond, VA 23298-0306; Phone: (804) 828-6938; Fax: (804) 828-4862; e-mail: wrsmith@vcu.edu