
Abstract
Pain is a frequent complaint of people living with sickle cell disease (SCD); however, the neurobiology of pain in SCD remains poorly understood. Whereas this pain has been thought to be primarily related to visceral and somatic tissue injury subsequent to vaso-occlusion events, emerging evidence from human and animal studies has suggested that a component of SCD pain may be related to neuropathic processes. Significant knowledge has been obtained from studies of molecular and neurobiological mechanisms leading to and maintaining neuropathic pain. Some of the most promising evidence has implicated major roles of protein kinase C and Ca2+/calmodulin-dependent protein kinase II, and their interaction with the N-methyl-D-aspartate receptors and the transient receptor potential vanilloid 1 receptor in the development of neuropathic pain. The latest evidence from our studies suggests that these pathways are important for SCD pain as well. Coupled with emerging animal models of SCD pain, we can now start to elucidate neurobiological mechanisms underlying pain in SCD, which may lead to better understanding and effective therapies.
Introduction
Pain and sickle cell disease (SCD) are so intimately intertwined, that African tribal words for the disease, spoken hundreds of years before Herrick described SCD in the Western literature,1 are onomatopoeic for pain. A century since Herrick's paper,1 the neurobiology of chronic pain in SCD remains poorly understood.
On the other hand, significant research effort has been made in elucidating the neurobiological mechanisms leading to other chronic pain conditions, including neuropathic pain. Neuropathic pain is defined as persistent pain resulting from damage to the peripheral or central nervous system (CNS) or abnormal communication within the nervous system.2 This syndrome is often manifested in patients suffering from diseases such as diabetes, herpes zoster, nerve traction or compression, complex regional pain syndrome, fibromyalgia, and AIDS, as well as in those with post-radiation pain. Many of these patients experience both persistent spontaneous pain and stimulus-evoked pain. The latter is characterized by both allodynia (pain elicited by normally innocuous, low-threshold stimuli) and hyperalgesia (enhanced pain response to noxious stimuli). Some recent evidence from our ongoing studies in SCD patients and in SCD transgenic models suggests the presence of neuropathic pain. The finding of neuropathic pain in SCD patients is contrary to the common belief that this pain is only nociceptive, and this may have a profound impact on our understanding and treatment of SCD pain. This article highlights some of the key advances in the study of molecular mechanisms underlying neuropathic pain that may be relevant to SCD pain, with an emphasis on signaling mechanisms involving protein kinases. Some other aspects of mechanisms relative to neuropathic pain, including glial activation, proinflammatory or pro-nociceptive cytokines, and chemokine receptors and transporters, can be found in several excellent reviews that have been published recently.3–5 These mechanisms are largely untested in SCD pain studies.
Neuropathic Pain in Adult SCD
Pain is a frequent complaint of people living with SCD. Nationwide epidemiological survey data indicate that over half of SCD patients have one to two episodes annually, and 1% of patients have more than 10 episodes each year.6 Hospital admissions for acute painful episodes have been reported to be a predictor of prognosis, and half of these hospitalized patients are readmitted within 1 month after discharge.7 A more recent study followed diary recordings of SCD patients for up to 6 months, and found that 55% of patients with SCD reported pain on more than half of the diary days and 29% on 95% of days.8 We followed 145 adult SCD patients who completed a computerized McGill Pain Questionnaire in an outpatient SCD specialty clinic. These subjects reported an average of 3.6 pain sites. Based on estimated normative scores on this questionnaire, we found that SCD pain was more severe than cancer pain or the pain of labor.40
Strikingly, SCD patients select verbal pain quality descriptors that are consistent with the presence of neuropathic pain. Over 90% of the 145 patients we studied selected neuropathic pain descriptors, and 59% reported continuous pain patterns. Our ongoing study using quantitative sensory testing is examining allodynia and hyperalgesia, the hallmarks of neuropathic pain in these patients. We also found long-lasting tactile allodynia and thermal (both cold and heat) hyperalgesia in SCD transgenic mice (Chen et al., unpublished observations).
Nociceptive Pain
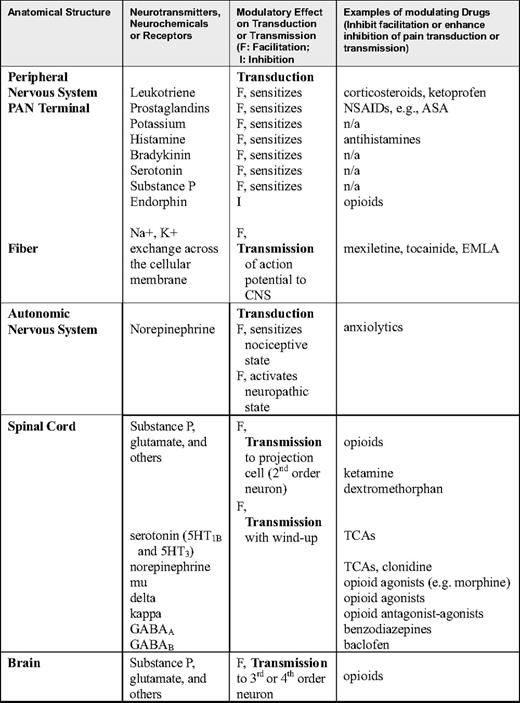
Primary afferent nociceptors (PAN), which sense thermal, mechanical, and chemical stimuli, are the peripheral nerve fibers of pseudounipolar sensory neurons that reside in the trigeminal ganglion (for the face) or the dorsal root ganglion (for the body). Sensory neurons receive peripheral pain stimuli from their target organs and transmit the nociceptive signals through their central projections that end in the spinal cord dorsal horns. Depending on the types of nociceptive nerve fibers and pain, different fibers terminate at different layers in the dorsal horn. Damage to visceral and somatic tissue produces a variety of chemicals that cause a neuronal action potential on the PAN. Some chemical by-products of cellular damage (e.g., bradykinin, serotonin, histamine, potassium ions, norepinephrine) stimulate the PAN, whereas others (e.g., leukotrienes, prostaglandins, substance P) sensitize the PAN to the effects of other mediators of neuronal transduction, which, if of sufficient magnitude, result in an action potential that is then transmitted to the spinal cord. Specifically, when a cell is damaged, phospholipids and other substances are liberated from the cell. The release of phospholipids initiates the arachidonic acid cascade through which 5-lipooxygenase and cyclooxygenase synthesize leukotrienes and prostaglandins, respectively. These events are shown in Figure 1.

Inhibition of the arachidonic acid pathway by several drugs. Steroids inhibit the production of arachidonic acid, thereby blocking the synthesis of leukotrienes and prostaglandins. Ketoprophen is believed to block the production of 5-lipooxygenase and cyclooxygenase. Aspirin and nonsteroidal anti-inflammatory drugs block the conversion of cyclo-oxygenase to prostaglandins and thromboxane A2. Trilisate blocks theproduction of prostagandins (PG) E2 and F2, but not I2 or thromboxane A2. (©2001 D.J. Wilkie, used with permission.)
Inhibition of the arachidonic acid pathway by several drugs. Steroids inhibit the production of arachidonic acid, thereby blocking the synthesis of leukotrienes and prostaglandins. Ketoprophen is believed to block the production of 5-lipooxygenase and cyclooxygenase. Aspirin and nonsteroidal anti-inflammatory drugs block the conversion of cyclo-oxygenase to prostaglandins and thromboxane A2. Trilisate blocks theproduction of prostagandins (PG) E2 and F2, but not I2 or thromboxane A2. (©2001 D.J. Wilkie, used with permission.)
Leukotrienes and prostaglandins sensitize the PAN to be activated by a smaller stimulus. For example, light pressure is not perceived as painful in normal conditions, but sometimes is sensed as pain (allodynia) if there is an inflammatory response with leukotrienes or prostaglandins facilitating the action potential in the PAN. In addition to the arachidonic cascade, many chemicals activate the PAN when they leak out of the cell or are released into the intracellular space as part of the inflammatory response. For example, potassium and histamine exude from damaged cells, and bradykinin is degraded from plasma kininogen, a component of inflammatory exudate. Other chemicals are released from platelets (serotonin) or mast cells (histamine). Sufficient concentrations of any of these chemicals around the PAN will cause the PAN to be transduced with the action potential and then transmitted to the spinal dorsal horn. These chemicals also act in combination to sensitize the PAN, enabling it to fire with a stimulus smaller than usual. Furthermore, if the PAN is transduced and an action potential is initiated, the PAN itself releases chemicals, one of which is substance P, which is stored in the distal terminals of the PAN and released through a retrograde process. In this way, substance P sensitizes the PAN, dilating nearby blood vessels, which leads to local edema and causes release of histamine from mast cells. In summary, tissue injury (e.g., that due to vaso-occlusive events or inflammation) results in the production and release of a number of chemicals around the PAN. These chemicals can sensitize or transduce the PAN directly (nociceptive pain) and through secondary processes (usually nociceptive pain unless neural tissue has been injured, resulting in neuropathic pain). These chemicals are commonly described as ingredients in the peripheral milieu surrounding the PAN. If any or all of these ingredients are eliminated from the peripheral milieu, then the PAN action potential may not be transmitted to the CNS. A number of non-opioid analgesics inhibit these chemicals and thereby remove peripheral milieu ingredients that contribute to PAN activation. Drugs that block the production or release of these chemicals can be powerful analgesics, and therefore constitute the first line of analgesic therapy (Table 1).
Central Sensitization and Neuropathic Pain
Significant CNS neuroplasticity occurs after neuronal damage, resulting in central sensitization.4,9 Central sensitization requires, in part, the activation of the N-methyl-D-aspartate (NMDA) receptors because the development of the sensitized state can be blocked by NMDA receptor antagonists.9 The NMDA receptor is a ligand-gated ion channel that is open when the receptor is bound to glutamate and the membrane is sufficiently depolarized. The latter is required for the dissociation of the Mg2+ that sits in the pore and blocks the channel at the resting state. The activated NMDA receptor is highly permeable to the influx of Ca2+. It has also been demonstrated that NMDA receptor antagonists (competitive or noncompetitive) administered prior to inflammatory pain or peripheral nerve injuries suppress or delay the onset of hyperalgesia and attenuate fully developed hyperalgesia when administered after the injury.10 Interestingly, magnesium has been reported to be beneficial for SCD pain.11,12 Activation of the NMDA receptor and subsequent activation of nitric oxide synthase (NOS) through Ca2+ influx and Ca2+/CaM-dependent pathway is well established.13 Inhibitors of NOS have been consistently shown to be effective in different chronic pain models.14–16 The NOS/NO pathway is particularly relevant for SCD. However, clinical applications of NOS inhibitors in SCD should proceed with caution, as NO may be both beneficial and harmful in SCD. Among the three isoforms of NOS, neuronal, endothelial, and inducible, neuronal NOS is the most important for pain. Thus, pharmacological intervention on pain in SCD should be focused on inhibiting this type of NOS.17 The mechanism of activation of the NMDA receptor complex during the development of neuropathic pain is uncertain. Persistent activation of the NMDA receptors through receptor phosphorylation has been proposed, because the NMDA receptor can be phosphorylated and activated by several protein kinases, including protein kinase C (PKC) and Ca2+/calmodulin-dependent protein kinase II (CaMKII). In addition, PKC can activate the NMDA receptor by reducing Mg2+ blockade.
PKC and Neuropathic Pain
PKC isoforms regulate the function of a variety of substrates by phosphorylation. This enzyme class is involved in diverse cellular processes, from cell differentiation and proliferation to long-term potentiation of neuronal activity. PKCs are classified into three groups based on their primary structure and cofactors for activation. The conventional PKC group includes PKCα, PKCβI, PKCβII, and PKCγ, which are activated by calcium and phosphatidylserine. In contrast, the novel PKCs, including PKCδ, PKCε, PKCη, and PKCθ, do not require calcium for activation. PKCζ and PKCι (human homolog of murine λ) belong to the class of atypical PKCs, which are activated by an as-yet-undefined mechanism. PKCs have been found to translocate to the membrane upon influx of Ca2+. Protein kinase inhibitors, including those relatively selective for PKCs, have been shown to block behavioral signs of pain in animal models. More specifically, studies have linked isoforms of PKC with pain. PKCγ immunoreactivity was reported to be increased after peripheral nerve injury and inflammation.18 In null mutants of PKCγ, nerve injury-induced allodynia and thermal hyperalgesia were largely absent.19 However, PKCγ is not the only isoform involved in neuropathic pain. Epinephrine- or acetic acid-induced hyperalgesia was markedly attenuated in mice lacking the PKCε gene.20 The NMDA receptor-mediated currents are potentiated by PKC and PKC activators.21 The activated NMDA receptors are permeable to Ca2+, which in turn can activate PKC. Therefore, a positive feed-forward loop between PKC and the NMDA receptors may exist (Figure 2), although such a mechanism has yet to be tested directly in neuropathic pain.

Positive feed-forward loops involving PKC and CaMKII in neuropathic pain. This is a simplified scheme demonstrating the persistent activation of PKC and CaMKII in neuropathic pain. Ca2+ influx through the activation of the NMDA receptors or TRPV1 leads to increased intracellular Ca2+ levels and subsequent activation of PKC and CaMKII. Both PKC and CaMKII can activate the NMDA receptors or TRPV1 via phosphorylation. Such feed-forward mechanisms may be one way for the sustained activation of PKC or CaMKII in neuropathic pain. It is likely that additional mechanisms can activate and/or sustain the activation of PKC and CaMKII, for example, by activating a Gq-coupled receptor or through Ca2+ release from the intracellular storage. It should be noted that PKC and CaMKII do not need to be co-expressed or simultaneously activated in a single cell. Whereas the NMDA receptors are expressed in the post-synaptic neurons, the TRPV1 may play a major role in pre-synaptic neurons. For downstream signal transduction, these protein kinases phosphorylate numerous effectors, including transcription factors, receptors, and ion channels.
Positive feed-forward loops involving PKC and CaMKII in neuropathic pain. This is a simplified scheme demonstrating the persistent activation of PKC and CaMKII in neuropathic pain. Ca2+ influx through the activation of the NMDA receptors or TRPV1 leads to increased intracellular Ca2+ levels and subsequent activation of PKC and CaMKII. Both PKC and CaMKII can activate the NMDA receptors or TRPV1 via phosphorylation. Such feed-forward mechanisms may be one way for the sustained activation of PKC or CaMKII in neuropathic pain. It is likely that additional mechanisms can activate and/or sustain the activation of PKC and CaMKII, for example, by activating a Gq-coupled receptor or through Ca2+ release from the intracellular storage. It should be noted that PKC and CaMKII do not need to be co-expressed or simultaneously activated in a single cell. Whereas the NMDA receptors are expressed in the post-synaptic neurons, the TRPV1 may play a major role in pre-synaptic neurons. For downstream signal transduction, these protein kinases phosphorylate numerous effectors, including transcription factors, receptors, and ion channels.
CaMKIIα and Pain
CaMKII is a multifunctional, CaM-activated serine/threonine protein kinase that is a key component of intracellular Ca2+-signaling pathways.22 Upon a significant increase in intracellular Ca2+ levels, calmodulin is activated by a conformational change after its binding to Ca2+ at the Ca2+-binding sites. This binding leads to the activation of CaMKII when the latter is bound with Ca2+/CaM. A key step in CaMKII activation is the autophosphorylation of threonine 286/287 upon the binding of Ca2+/CaM. The autophosphorylation renders the kinase fully active, and it can remain active even after the Ca2+ has subsided.
CaMKII is ubiquitously distributed in the CNS and accounts for approximately 1% to 2% of total brain protein mass. In the CNS, CaMKII is involved in a variety of Ca2+-mediated cellular processes, including biosynthesis of neurotransmitters, hormone secretion, neurotransmitter release, gene expression, and neuronal plasticity.23 In this kinase family, CaMKIIα is the most extensively studied isoform that has been found to regulate synapse transduction by phosphorylating ion channels and signal transduction molecules at post-synaptic density.22
Both the release of Ca2+ from intracellular storages and Ca2+ influx as a result of receptor or ion channel activation can activate calmodulin and CaMKII. Activation of the transient receptor potential vanilloid 1 receptor (TRPV1) or the NMDA receptors will both lead to Ca2+ influx and CaMKII activation. The unique interaction between CaMKII and NMDA receptors is intriguing and may serve as a mechanism for long-lasting plasticity in neuropathic pain. CaMKII phosphorylates the NMDA receptor, leading to receptor activation. Ca2+ influx through the activation of the NMDA receptors activates CaM and leads to the autophosphorylation and subsequent full activation of CaMKII. Therefore, a positive feed-forward loop keeps the pathways active after the original Ca2+ signaling has subsided or disappeared. Indeed, inflammatory pain and nerve injury have been shown to increase the levels of glutamate in the spinal dorsal horn that were blocked by NMDA receptor antagonists.10 Overexpression of the NR2B subunit of the NMDA receptor causes increased mechanical allodynia in conditions with inflammatory pain, whereas inhibition of NR2B24 or knockdown of spinal NMDA receptors with antisense oligonucleotides25 prevents the expression of chemically induced hyperalgesia. Therefore, CaMKII may work in concert with the NMDA receptors in the development and maintenance of the neural events leading to hyperalgesia.
In the spinal cord and the primary afferent nerves, CaMKIIα is specifically expressed in the superficial laminae of the spinal dorsal horn and in the small- to medium-diameter primary sensory neurons in dorsal root ganglia, where nociceptive signals are transmitted and processed.26,27 Willis et al. reported that CaMKIIα activity was increased significantly in the spinal cord within minutes after an intradermal injection of capsaicin.28 In addition, spinally administered CaMKII inhibitor KN93, but not inactive analog KN92, inhibited the enhancement of responses of spinal nociceptive neurons and changes in exploratory behavior evoked by capsaicin injection.28 In rat trigeminal ganglion neurons, Hargreaves et al. found that capsaicin increased CaMKII activity in TRPV1-positive neurons.29 In these trigeminal ganglion neurons, capsaicin- or n-arachidonoyl-dopamine-evoked calcitonin gene-related peptide release was inhibited by KN93.29 These data suggest that there is a functional interaction between CaMKII and TRPV1. Mechanistically, CaMKII phosphorylates TRPV1, thereby regulating TRPV1 activity. TRPV1 regulates the Ca2+ influx and intracellular Ca2+ levels, thereby affecting CaMKII activity.30 Therefore, there may be a functional interaction between CaMKII and TRPV1, similar to that between CaMKII and the NMDA receptor complex (Figure 2).
The role of CaMKIIα in persistent inflammatory or neuropathic pain has also been suggested. One group found that CaMKII inhibitors at very low doses reversed chronic nerve constriction injury-induced neuropathic pain in mice, although the exact experimental details were not provided.25 In other studies, investigators reported mixed effects in that KN93 prevented, but did not reverse, thermal hyperalgesia and mechanical allodynia following constriction injury-induced neuropathic pain.10 We have reported a critical role of CaMKII in complete Freund's adjuvant-induced hyperalgesia pain,31 spinal nerve ligation-induced neuropathic pain behaviors,32 and opioid-induced hyperalgesia.33 In summary, CaMKIIα appears to be a critical regulator of central sensitization, leading to many, if not all, types of chronic pain.
Neurobiological Mechanisms of SCD Pain
Early studies on the molecular mechanisms of neuropathic pain were hindered by the lack of suitable models until several experimental neuropathic injury models were developed in rodents.36 Since then, these models have been used extensively to examine the neuroanatomical and neurochemical changes in the spinal cord and other CNS regions that contribute to the development and maintenance of neuropathic pain. We have seen similar limitations in the study of pain in SCD. However, SCD transgenic mice models have now emerged as more suitable models for the disease and for the study of SCD pain.
The first-generation SCD mouse model was transgenic, expressing human α and βS globin transgenes without deletion of endogenous mouse α and β genes.37 Limited red cell sickling was observed in these mice in vivo.38 In order to enhance red cell sickling, a β-globin gene with three mutations (designated SAD) was introduced.39 Additional mouse models have been produced to yield models with varying severity hemolytic anemia and other symptoms of SCD.37 A major breakthrough came when SCD mouse lines were made to carry exclusively human globins by “knocking out” mouse globin genes.40–42 Among these mouse models of SCD, Berkeley sickle-cell transgenic mice express exclusively human sickle hemoglobin and have a phenotype that closely mimics many features of severe SCD in humans, including severe hemolytic anemia, irreversibly sickled red cells, increased rigidity of erythrocytes, and extensive multiple organ damage.43 Additional similarities to human disease include vascular ectasia, intravascular hemolysis, exuberant hematopoiesis, cardiomegaly, glomerulosclerosis, visceral congestion, hemorrhages, multi-organ infarcts, pyknotic neurons, progressive siderosis, gallstones, and priapism.44 Littermate mice that are heterozygous for Hbb gene do not develop hemolytic anemia or other SCD characteristics.44
The NY1DD mice, having mild SCD pathology and no hemolysis, have been studied for pain behaviors. In a thermal nociception test in which radiant heat was applied to the dorsal side of the tail, the latency to a rapid tail-flick response was compared between NY1DD mice and C57Bl/6 control mice. At 10 weeks of age, NY1DD mice showed a more rapid tail-flick response (p < 0.0001), suggesting the presence of thermal hyperalgesia. At 6 weeks of age, however, the latencies exhibited by the two groups of mice were not different. These data suggest that NY1DD sickle cell mice have an age-dependent onset of thermal hyperalgesia.45
In our ongoing studies of Berkeley sickle cell mice, we have employed assays that are predominately designed to detect neuropathic pain, including cold allodynia, heat hyperalgesia, and mechanical allodynia. We observed a significantly enhanced pain response and decreased nociception threshold that persisted for at least 4 months in Berkeley sickle cell mice compared with littermate non-sickle-cell mice (Chen et al., unpublished observations). These data are in agreement with our clinical findings of the presence of neuropathic pain in adult humans with SCD.46 Moreover, we found that PKC and CaMKII activation were significantly increased in the spinal dorsal horn neurons, which was similar to what we have observed in other chronic pain states.31–33
Summary
Intriguing evidence from adults with SCD and transgenic sickle cell mice challenges the existing assumptions about pain in SCD. Whereas pain with SCD has been thought to be related to visceral and somatic tissue injury subsequent to vaso-occlusion events, another component of the pain may be related to neuropathic processes. Some of the mechanisms related to neuropathic pain involve CaMKII and PKC, which may be relevant to the pain of SCD. Our group is characterizing the mechanisms of pain in Berkeley transgenic sickle cell mice. We are also characterizing thermal and mechanical allodynia and hyperalgesia in adult patients with SCD. Timely with the 100-year anniversary of scientific documentation of SCD, the insights from these studies will offer important evidence about the role of neuropathic pain in SCD.
Acknowledgments
This publication was made possible by National Institutes of Health grants R01 HL098141, R01 HL078536, and K07 AT003647. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health, National Heart Lung and Blood Institute, or National Center for Complimentary and Alternative Medicine. The final peer-reviewed manuscript is subject to the National Institutes of Health Public Access Policy.
Disclosures
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Off-label drug use: None disclosed.
Correspondence
Zaijie Jim Wang, Department of Biopharmaceutical Sciences, University of Illinois at Chicago, MC865, Room 335, 833 S. Wood Street, Chicago, IL 60612; Phone: 312-355-1429; Fax: 312-413-7792; e-mail: zjwang@uic.edu
