

Key Points
Germline heterozygous mutations in IKZF2 cause a novel syndrome associated with immunodeficiency and profound immuno-dysregulation.
Germline heterozyogous mutations in IKZF2 disrupt interaction with the NuRD complex.

Abstract
Helios, encoded by IKZF2, is a member of the Ikaros family of transcription factors with pivotal roles in T-follicular helper, NK- and T-regulatory cell physiology. Somatic IKZF2 mutations are frequently found in lymphoid malignancies. Although germline mutations in IKZF1 and IKZF3 encoding Ikaros and Aiolos have recently been identified in patients with phenotypically similar immunodeficiency syndromes, the effect of germline mutations in IKZF2 on human hematopoiesis and immunity remains enigmatic. We identified germline IKZF2 mutations (one nonsense (p.R291X)- and 4 distinct missense variants) in six patients with systemic lupus erythematosus, immune thrombocytopenia or EBV-associated hemophagocytic lymphohistiocytosis. Patients exhibited hypogammaglobulinemia, decreased number of T-follicular helper and NK cells. Single-cell RNA sequencing of PBMCs from the patient carrying the R291X variant revealed upregulation of proinflammatory genes associated with T-cell receptor activation and T-cell exhaustion. Functional assays revealed the inability of HeliosR291X to homodimerize and bind target DNA as dimers. Moreover, proteomic analysis by proximity-dependent Biotin Identification revealed aberrant interaction of 3/5 Helios mutants with core components of the NuRD complex conveying HELIOS-mediated epigenetic and transcriptional dysregulation.
Introduction
Helios (encoded by IKZF2) is a member of the Ikaros transcription factor family characterized by 4 highly conserved N-terminal C2H2 zinc finger domains involved in DNA binding and 2 C-terminal C2H2 zinc fingers required for homodimeric and heterodimeric protein interactions with other family members, such as Ikaros or Aiolos.1 Helios has been shown to control lymphocyte development and T follicular helper (Tfh)– and natural killer (NK)–cell differentiation and to maintain the suppressive function of regulatory T (Treg) cells and is frequently deleted in hypodiploid B-cell acute lymphoblastic leukemia (B-ALL).2-6
Monoallelic germline mutations in Ikaros (IKZF1) and Aiolos (IKZF3) have been associated with combined immunodeficiency, common variable immunodeficiency, systemic lupus erythematosus (SLE), immune thrombocytopenia (ITP) as well as leukemia predisposition, with incomplete penetrance.7-12 Here, we identify patients with germline monoallelic mutations in IKZF2 presenting with features of immune dysregulation, including ITP, SLE, and susceptibility to Epstein-Barr virus–driven complications. We determine the detrimental effects of these mutations on Helios function, illustrating the key role of Helios in maintaining immune homeostasis.
Methods
Single-cell RNA-sequencing
Single-cell RNA-sequencing (scRNAseq) was performed on peripheral blood mononuclear cells (PBMCs) from the index patient (R291X) and 4 controls using the 10× Genomics Chromium Controller with the Chromium Single Cell 3′ Reagent Kit (v3 Chemistry) following the manufacturer’s instructions. After quality control, libraries were sequenced on the Illumina HiSeq 4000 platform in 2 × 75 bp paired-end mode. Supplemental Table 1 includes an overview of sequenced samples. More details are provided in the data supplement.
Interaction proteomics
Proximity-dependent Biotin Identification (BioID) technique, coupled with liquid chromatography–mass spectrometry was performed for Helios wild type and all variants according to Liu et al.13 More details are provided in the data supplement.
Results and discussion
Clinical phenotype and identification of IKZF2 variant
We studied a 16-year-old girl born to healthy nonconsanguineous parents (Figure 1A) who at 10 years of age developed skin lesions, photosensitivity, polyarthritis, and mild ITP with positive antiplatelet, anti-DNA, and antinuclear autoantibodies, prompting a diagnosis of SLE. The patient was in clinical remission upon treatment with hydroxychloroquine and nonsteroidal anti-inflammatory drugs (supplemental Table 1).
![Identification of a heterozygous variant in IKZF2 and effects on Helios function and transcription. (A) Family pedigree of the patient under study. The 16-year-old girl (A1) is the only affected member of the family. (B) Illustration of the Helios transcription factor, with 4 N-terminal zinc fingers responsible for DNA binding to the consensus sequence and 2 C-terminal zinc fingers that form the homo/heterodimerization domain. Location of the variant (transcript ID ENST00000434687.5; c.871C>T, p.Arg291Ter, rs146574423) is shown (red arrow). (C) Nuclear extracts from 293T cells transfected with wild-type (WT) or the mutant were prepared, and electrophoretic mobility shift assay was performed using the IK-BS1 probe, an Ikaros consensus-binding sequence. Representative image of 7 independent experiments. In the empty vector (EV) and WT lanes, 2 faint bands are visible, the top being DNA complexes, the bottom free probe. The lower band (red asterisk) for Helios R291X, the size of which coincides with that of the DNA complex band seen in the EV and WT lanes, is the monomer binding to the DNA probe, indicating that this variant cannot bind DNA as a homodimer. (D) Immunofluorescence staining of NIH3T3 cells transfected with Helios WT or Helios R291X using an anti-Helios antibody, showing the formation of foci at pericentromeric heterochromatin regions. Graph on the bottom shows the quantification of number of foci per cell. Results represent 3 independent experiments. For statistical analysis, an unpaired t test was performed. (E) Coimmunoprecipitation experiments after cotransfection of HEK293T cells with Streptavidin (Strep)-HA-Helios WT, Strep-HA-Helios R291X, FLAG-Helios WT, or an EV. Immunoprecipitations (IPs) were performed with Strep beads, and western blot analysis was performed by running both the (IP) and whole-cell lysate (input) on a gel and blotting with HA, FLAG, Helios, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies. Results are representative of 3 to 5 independent experiments. The expected size of untagged Helios was 60 kDa; the observed size increase is explained by addition of amino acids by the protein tag, with the Strep-HA tag being substantially longer than the FLAG tag. The sizes of the truncated variant R291X tagged with either FLAG or with Strep-HA were 45 and 55 KDa, respectively. (F) The top panel shows a low-dimensional projection (uniform manifold approximation and projection [UMAP] plot) of the combined scRNA-seq data set comprising 23 946 cells from the patient (P) and 4 controls. Numbers indicate clusters (graph-based clustering), and colors correspond to cell type (curation based on supplemental Figure 3A). The bottom panel shows the same projection, with colors indicating the distribution of patient cells (blue) within the clusters compared with controls (gray). (G) Bar plot showing the distribution of cells from each individual across the clusters defined in panel F. (H) Heatmap showing differentially expressed genes in clusters 2a and 2b, corresponding to T cells and T and NK cells, respectively. cDC, conventional dendritic cell; DAPI, 4′,6-diamidino-2-phenylindole; pDC, plasmacytoid dendritic cell. Scale bar: 25 µm.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/7/10.1182_bloodadvances.2021006367/7/m_advancesadv2021006367f1.png?Expires=1764956741&Signature=Qh3kF8QSjNtvzAe0Vm6jafMG75FIrf-jov35hWPqe~pzdZAfcA518nbR9cJffDKB9Qx5EBs4V0HgNdIJ54hQwoPHem1y21KwuOOea9Ja1k4Q2l5yxppWiurL8WdvLy8VMqkMZZx9qD5IUq6nssETNe3G15ggwepvuzNtxMN8s7qPJlIhy~3bYC1rK0GxKC1fmUKYBtsuBKLZjWnW0x-nSe3hdgTNorm8cFcJhQOVp0JZG8-QrKZ4qhvLRyg3Jk8IdabeOQuJxFJwrD0ZCq7pJyMvA8vYTerUPOF84MeuRDgzx0s2Y0a4G584V7bXELChiPKteVNRU39PQhWwwcE5dg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Identification of a heterozygous variant in IKZF2 and effects on Helios function and transcription. (A) Family pedigree of the patient under study. The 16-year-old girl (A1) is the only affected member of the family. (B) Illustration of the Helios transcription factor, with 4 N-terminal zinc fingers responsible for DNA binding to the consensus sequence and 2 C-terminal zinc fingers that form the homo/heterodimerization domain. Location of the variant (transcript ID ENST00000434687.5; c.871C>T, p.Arg291Ter, rs146574423) is shown (red arrow). (C) Nuclear extracts from 293T cells transfected with wild-type (WT) or the mutant were prepared, and electrophoretic mobility shift assay was performed using the IK-BS1 probe, an Ikaros consensus-binding sequence. Representative image of 7 independent experiments. In the empty vector (EV) and WT lanes, 2 faint bands are visible, the top being DNA complexes, the bottom free probe. The lower band (red asterisk) for Helios R291X, the size of which coincides with that of the DNA complex band seen in the EV and WT lanes, is the monomer binding to the DNA probe, indicating that this variant cannot bind DNA as a homodimer. (D) Immunofluorescence staining of NIH3T3 cells transfected with Helios WT or Helios R291X using an anti-Helios antibody, showing the formation of foci at pericentromeric heterochromatin regions. Graph on the bottom shows the quantification of number of foci per cell. Results represent 3 independent experiments. For statistical analysis, an unpaired t test was performed. (E) Coimmunoprecipitation experiments after cotransfection of HEK293T cells with Streptavidin (Strep)-HA-Helios WT, Strep-HA-Helios R291X, FLAG-Helios WT, or an EV. Immunoprecipitations (IPs) were performed with Strep beads, and western blot analysis was performed by running both the (IP) and whole-cell lysate (input) on a gel and blotting with HA, FLAG, Helios, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies. Results are representative of 3 to 5 independent experiments. The expected size of untagged Helios was 60 kDa; the observed size increase is explained by addition of amino acids by the protein tag, with the Strep-HA tag being substantially longer than the FLAG tag. The sizes of the truncated variant R291X tagged with either FLAG or with Strep-HA were 45 and 55 KDa, respectively. (F) The top panel shows a low-dimensional projection (uniform manifold approximation and projection [UMAP] plot) of the combined scRNA-seq data set comprising 23 946 cells from the patient (P) and 4 controls. Numbers indicate clusters (graph-based clustering), and colors correspond to cell type (curation based on supplemental Figure 3A). The bottom panel shows the same projection, with colors indicating the distribution of patient cells (blue) within the clusters compared with controls (gray). (G) Bar plot showing the distribution of cells from each individual across the clusters defined in panel F. (H) Heatmap showing differentially expressed genes in clusters 2a and 2b, corresponding to T cells and T and NK cells, respectively. cDC, conventional dendritic cell; DAPI, 4′,6-diamidino-2-phenylindole; pDC, plasmacytoid dendritic cell. Scale bar: 25 µm.
Identification of a heterozygous variant in IKZF2 and effects on Helios function and transcription. (A) Family pedigree of the patient under study. The 16-year-old girl (A1) is the only affected member of the family. (B) Illustration of the Helios transcription factor, with 4 N-terminal zinc fingers responsible for DNA binding to the consensus sequence and 2 C-terminal zinc fingers that form the homo/heterodimerization domain. Location of the variant (transcript ID ENST00000434687.5; c.871C>T, p.Arg291Ter, rs146574423) is shown (red arrow). (C) Nuclear extracts from 293T cells transfected with wild-type (WT) or the mutant were prepared, and electrophoretic mobility shift assay was performed using the IK-BS1 probe, an Ikaros consensus-binding sequence. Representative image of 7 independent experiments. In the empty vector (EV) and WT lanes, 2 faint bands are visible, the top being DNA complexes, the bottom free probe. The lower band (red asterisk) for Helios R291X, the size of which coincides with that of the DNA complex band seen in the EV and WT lanes, is the monomer binding to the DNA probe, indicating that this variant cannot bind DNA as a homodimer. (D) Immunofluorescence staining of NIH3T3 cells transfected with Helios WT or Helios R291X using an anti-Helios antibody, showing the formation of foci at pericentromeric heterochromatin regions. Graph on the bottom shows the quantification of number of foci per cell. Results represent 3 independent experiments. For statistical analysis, an unpaired t test was performed. (E) Coimmunoprecipitation experiments after cotransfection of HEK293T cells with Streptavidin (Strep)-HA-Helios WT, Strep-HA-Helios R291X, FLAG-Helios WT, or an EV. Immunoprecipitations (IPs) were performed with Strep beads, and western blot analysis was performed by running both the (IP) and whole-cell lysate (input) on a gel and blotting with HA, FLAG, Helios, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies. Results are representative of 3 to 5 independent experiments. The expected size of untagged Helios was 60 kDa; the observed size increase is explained by addition of amino acids by the protein tag, with the Strep-HA tag being substantially longer than the FLAG tag. The sizes of the truncated variant R291X tagged with either FLAG or with Strep-HA were 45 and 55 KDa, respectively. (F) The top panel shows a low-dimensional projection (uniform manifold approximation and projection [UMAP] plot) of the combined scRNA-seq data set comprising 23 946 cells from the patient (P) and 4 controls. Numbers indicate clusters (graph-based clustering), and colors correspond to cell type (curation based on supplemental Figure 3A). The bottom panel shows the same projection, with colors indicating the distribution of patient cells (blue) within the clusters compared with controls (gray). (G) Bar plot showing the distribution of cells from each individual across the clusters defined in panel F. (H) Heatmap showing differentially expressed genes in clusters 2a and 2b, corresponding to T cells and T and NK cells, respectively. cDC, conventional dendritic cell; DAPI, 4′,6-diamidino-2-phenylindole; pDC, plasmacytoid dendritic cell. Scale bar: 25 µm.
To investigate a potential monogenic etiology of the disease, we performed whole-exome sequencing and identified a paternally inherited heterozygous variant in IKZF2 located after the DNA-binding domain that results in a premature stop codon in exon 8 (c.871C>T, p.Arg291Ter; Figure 1B). Stringent filtering revealed that the variant was ultrarare (Genome Aggregation Database [gnomAD] minor allele frequency, 4 × 10−6) and predicted to be deleterious (Combined Annotation Dependent Depletion score of 38; supplemental Table 2). IKZF2 is annotated with a high probability of loss of function intolerance score of 0.99 (gnomAD version 2.1.1), and compared with the probability of loss-of-function intolerance of other genes causing inborn errors of immunity (IEIs) in a haploinsufficient state, it ranks 4 of 13 (supplemental Figure 1A), indicating strong susceptibility to haploinsufficiency.14 Interestingly, the patient’s asymptomatic father carried the same variant, indicating incomplete penetrance,15 a feature that has also been reported in other IEIs, including germline heterozygous IKZF1 mutations.16
Functional and biochemical assessment of Helios R291X
We assessed the effects of the Helios R291X mutation on the ability to bind DNA and dimerize. An electrophoretic mobility shift assay using lysates of HEK293T cells overexpressing either Helios wild type or Helios R291X showed that the R291X mutation abolished dimerization and consequently DNA-homodimer complex formation (Figure 1C). Consistently, Helios R291X failed to bind to pericentromeric heterochromatin regions or localize to nuclear foci (Figure 1D), confirming reduced ability to bind target DNA as dimers. Additionally, coimmunoprecipitation experiments showed that Helios R291X was neither able to homodimerize (Figure 1E) nor to form heterodimers with Ikaros or Aiolos (supplemental Figure 2).
Alterations in the transcriptional state of patient immune cells
We next addressed whether the impaired DNA binding of the mutant as dimers and its inability to form homo- or heterodimers translated into changes in the transcriptional landscape. We performed droplet-based scRNAseq on patient and healthy donor PBMCs. Low-dimensional projection using uniform manifold approximation and projections and graph-based clustering confirmed the presence of expected PBMC populations17 (Figure 1F-G; supplemental Figure 3A; supplemental Table 3). We observed no overt differences of patient cells in the B-cell cluster. However, we observed a decrease of patient cells in cluster 2b, corresponding to cells expressing genetic characteristic of NK cells and central memory T cells. We also observed an increase in the number of cells in cluster 1a, corresponding to CD14+ classical monocytes (Figure 1F-G). Differential gene expression analysis between patient and control cells (Figure 1H; supplemental Figure 3B) revealed an upregulation of genes associated with inflammation, indicating an activated and effector state in both monocytes and T cells. For instance, in clusters 2a and 2b, we found transcriptional upregulation of genes induced upon T-cell receptor activation, such as JUNB and HSPA5.18,19 Moreover, upregulation of DUSP2 in effector T cells is associated with a conversion to exhausted T cells20 and promotes proinflammatory Th17-cell differentiation.21
Flow cytometric analysis of patient immune cells
Our scRNAseq-based observations were further substantiated by flow cytometry (Figure 2A-F; supplemental Table 4); whereas absolute numbers of B cells in the patient were unremarkable, those of CD27+IgD− class-switched B cells were borderline low (Figure 2B). Within the T-cell fraction, we observed a reduction in CD4+ T cells, whereas CD8+ cell numbers were normal (supplemental Table 4). Because Tfh cells expressed Helios in an alum vaccination system in the OTII mouse model and a Heliosfl/fl × Foxp3Cre conditional knockout of Helios produced increased propensities of splenic Tfh cells, we examined circulating Tfh-like cells, which, in contrast to the mouse model, were reduced in the patient (Figure 2C).22,23 T-cell maturation was normal (supplemental Table 4). Reminiscent of previous findings,5 our scRNAseq data also showed that Helios is highly expressed in Treg and NK cells (supplemental Figure 3C). Consistently, we observed reduced CD16+CD56dim NK cells in the patient (Figure 2D). However, CD127−CD25+FOXP3+ Treg cells were normal in quantity (Figure 2F), but Helios expression was reduced in these cells (Figure 2G), suggesting a potential functional Treg impairment.
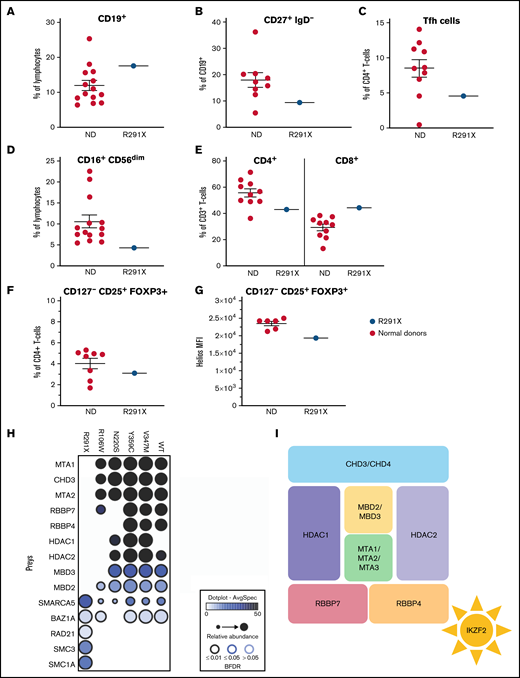
Aberrant immune phenotype of the index patient and interactome in the patient cohort. (A-F) Graphs showing the frequency of B cells (CD3−CD19+) within lymphocytes (A) and class-switched (CD27+IgD−) B cells within CD3−CD19+ cells (B). (C) Tfh cells (CXCR5+CD45RA−) within CD3+CD4+ cells. (D) NK cells (CD16+CD56dim) within lymphocytes. (E) CD4+ and CD8+ cells within total (CD3+) T cells. (F) Proportion of Treg cells (CD127−CD25+FOXP3+) within CD3+CD4+ cells in patients and controls. (G) HELIOS mean fluorescence intensity (MFI) within CD127−CD25+FOXP3+ Treg cells. The Helios antibody used recognizes all Helios variants. (H) Dot plot representation of high-confidence Biotin Identification interactors that are known components of the nucleosome remodeling histone deacetylase (NuRD) complex of wild-type (WT) and mutant HELIOS. Dot size indicates relative abundance, dot fill color indicates average spectral count, and dot border color indicates statistical significance. (I) Schematic description of the NuRD complex. BFDR, Bayesian false discovery rate; ND, normal donor.
Aberrant immune phenotype of the index patient and interactome in the patient cohort. (A-F) Graphs showing the frequency of B cells (CD3−CD19+) within lymphocytes (A) and class-switched (CD27+IgD−) B cells within CD3−CD19+ cells (B). (C) Tfh cells (CXCR5+CD45RA−) within CD3+CD4+ cells. (D) NK cells (CD16+CD56dim) within lymphocytes. (E) CD4+ and CD8+ cells within total (CD3+) T cells. (F) Proportion of Treg cells (CD127−CD25+FOXP3+) within CD3+CD4+ cells in patients and controls. (G) HELIOS mean fluorescence intensity (MFI) within CD127−CD25+FOXP3+ Treg cells. The Helios antibody used recognizes all Helios variants. (H) Dot plot representation of high-confidence Biotin Identification interactors that are known components of the nucleosome remodeling histone deacetylase (NuRD) complex of wild-type (WT) and mutant HELIOS. Dot size indicates relative abundance, dot fill color indicates average spectral count, and dot border color indicates statistical significance. (I) Schematic description of the NuRD complex. BFDR, Bayesian false discovery rate; ND, normal donor.
Identification of additional patients with Helios mutation
Stimulated by the effect of the Helios R291X variant, we revisited our database of patients with IEIs. We identified 5 additional patients from 4 nonconsanguineous families carrying 4 different rare (gnomAD allele frequency <0.001) heterozygous missense variants in IKZF2, which were predicted to be damaging (c.1076A>G, p.Y359C; c.1038G>A, p.V347M; c.316C>T, p.R106W; and c.659A>G, p.N220S, respectively; supplemental Figures 1B-C). Intriguingly, these patients presented with profound immune dysregulation, manifesting in SLE or susceptibility to Epstein-Barr virus–associated HLH, similar to the clinical phenotype of the Helios R291X patient (supplemental Table 2 and data supplement for detailed clinical information). Importantly, in family D, the Helios R106W variant was also present in 2 family members who were asymptomatic, indicating incomplete penetrance.
Consistently, immunophenotypic analysis revealed similarities with the Helios R291X patient, including decreased numbers of Tfh and NK cells. Regarding the B-cell compartment, reduced percentages of class-switched memory B cells were present in Helios Y359C and Helios N220S patients, in line with the borderline reduction observed in the index patient, whereas Helios V347M and Helios R106W patients displayed hypogammaglobulinemia (supplemental Table 2; supplemental Figure 4). As in the Helios R291X patient, T-cell maturation, number of CD127+CD25+FOXP3+ Treg cells, and FOXP3 expression of CD25+FOXP3+Helios+ Treg cells were normal in the patients (supplemental Figure 4).
Previous studies in other germline–encoded defects in IKZF1 have illustrated a plethora of phenotypes dependent on specific locations of mutations.24 Similarly, we observed that, unlike the Helios R291X variant, the other 4 variants of the IKZF2 gene, except R106W, showed no overt defects in their ability to bind to pericentromeric heterochromatin regions in DNA (supplemental Figure 5) or form homodimers (supplemental Figure 6) or heterodimers with other Ikaros family members (supplemental Figure 2). Intrigued by the observation that all the variants lay outside the DNA and homo- and heterodimerization domains (supplemental Figure 1C), we hypothesized that the interaction with additional binding partners could be affected. Therefore, we sought to globally probe Helios interactions and identify which of those are consistently aberrant in all Helios variants. We used a proximity-dependent BioID technique, coupled with liquid chromatography–mass spectrometry13 (Figure 2H; supplemental Figures 7 and 8). We found a decreased interaction in 3 out of 5 variants (p.R291X, p.R106W, and p.N220S) with different components of the NuRD complex, such as CHD3, MTA1/2, or RBBP4/7 (Figure 2I). The NuRD complex has been implicated in transcriptional repression and activation and reported to be recruited by Helios to specific foci to regulate transcription.25 In this regard, it has been shown that inactivation of the NuRD complex in mice is associated with SLE-like autoimmune disease.26 Other chromatin remodelers that have been shown to function in close equilibrium with the NuRD complex, such as the SWI/SNF complex (SMARCA5) or the cohesin complex (RAD21, SMC3, and SMC1A), were also perturbed in at least 4 out of 5 of the patients (Figure 2H).27 This indicates that the balance of these activities is disrupted in these patients, ultimately resulting in transcription dysregulation. In line with these considerations, during the revision process of this manuscript, Hetmäki et al28 published their findings on 2 siblings carrying a germline heterozygous truncating variant in IKZF2. This variant also prevented its binding to members of the NuRD complex, further supporting the critical role of this complex in Helios function and activity.28 Collectively, additional studies identifying more patients will enable the definition of the phenotypic spectrum of germline heterozygous mutations in IKZF2, along with the identification of potential genotype-phenotype correlations.
Germline mutations in IKZF1 and IKZF3 predispose to B-ALL and B-cell lymphoma, respectively.12,29 Dysfunctional Helios has been implicated in sporadic hematopoietic malignancies; in adult T-cell lymphoma, deletions leading to aberrant splicing have been observed.30 Similar short isoforms lead to T-cell lymphoma in mice as a result of a dominant-negative effect with loss of DNA binding activity of the mutant protein.31 Furthermore, deletions affecting Helios are a hallmark of low hypodiploid B-ALL observed in 52.9% of patients.6 Although we did not observe malignancy in our cohort of patients, future studies will need to determine whether such IKZF2 germline mutations confer an increased risk of malignancy and whether close monitoring and potential preemptive therapy may be indicated.
Acknowledgments
The authors thank the patients and their family members for their participation in our study.
This study was supported by the European Research Council (ERC) Consolidator Grant iDysChart (ERC grant agreement 820074) (to K.B.); a doctoral fellowship program (“Cell Communication in Health and Disease”; co-financed by the Austrian Science Fund and the Medical University of Vienna) (K.B.); an Austrian Science Fund Hertha Firnberg fellowship (T934-B30) (B.H.); the Intramural Research Program, Clinical Center, US National Institutes of Health (S.D.R.); and the Academy of Finland (288475 and 294173), the Sigrid Juselius Foundation, the Finnish Cancer Institute, and Biocenter Finland (M.V.). F.R.-L. was supported by INSERM and by government grants from the Agence National de la Recherche as part of the Investment for the Future program (ANR-10-IAHU-01 and ANR-18-RHUS-0010), the Centre de Référence Déficits Immunitaires Héréditaires, the Agence National de la Recherche (ANR-14-CE14-0026-01 and ANR-18-CE17-0001), and the Fondation pour la Recherche Médicale (Equipe FRM EQU202103012670). J.H. was a recipient of an INSERM poste d’accueil program and an Institut Imagine PhD fellowship program supported by the Fondation Bettencourt Schueller.
Authorship
Contribution: T.S. performed most of the experiments; T.S. and D.M. collected and interpreted clinical, laboratory, and experimental data and prepared most of the figures and tables; T.S., H.S.K., B.H., and S.G. performed the biochemical experiments; M.R.S., under the supervision of F.H., analyzed single-cell RNA sequencing (scRNA-seq) data and prepared figures and tables; L.M.G., under the supervision of M.V., performed and analyzed Biotin Identification experiments and prepared figures; O.Y.P. performed and analyzed immunofluorescence microscopy experiments; J.H., D.B., M.P.T., T.A.F., F.R.-B., N.C.C., P.Q., and N. Romberg provided patient care, collected samples, and analyzed clinical and laboratory data; S.K.B. and N. Rezaei assisted in the interpretation of clinical data; T.S., D.M., and I.C. with help from M.R.S. and F.H. interpreted scRNAseq data with help from S.Z.; M.H. and D.M. interpreted population genetics data; R.J.H. assisted in the interpretation of genetic findings; S.L. and F.R.-L. analyzed genetic data, collected and interpreted laboratory data, and provided valuable input; I.C. interpreted biochemical experiments and prepared selected figures and tables; S.D.R. and H.S.K. provided the resources for vital experiments and provided valuable input for the study; S.D.R. and K.B. provided laboratory resources and data interpretation; S.G.T. assisted with statistical analysis; K.B. conceptualized and coordinated, and took overall responsibility for the study; T.S., D.M., I.C., S.G., and K.B. wrote the manuscript with help from all authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for B.H. is Institute of Pharmacology, Johannes Kepler University Linz, Linz, Austria.
Correspondence: Kaan Boztug, St. Anna Children’s Cancer Research Institute (CCRI) and Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases, Zimmermannplatz 10, A-1090 Vienna, Austria; e-mail: kaan.boztug@ccri.at or kaan.boztug@rud.lbg.ac.at.

