

TO THE EDITOR:
The COVID-19 infection is caused by a novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).1 The SARS-CoV-2 virus contains various nonstructural proteins and 4 major structural proteins: surface-exposed spike (S), membrane, envelope, and internal nucleocapsid (N) proteins.1,2 The S fusion protein consists of the S1 and S2 components, and the virus enters cells, such as pneumocytes in the lung,3 via binding of the receptor-binding domain (RBD) within the S1 protein4 to the angiotensin-converting enzyme-2 receptor.2,5
Patients with hematologic malignancies are at increased risk of morbidity and mortality from COVID-19 disease.6 This is likely a result of a combination of immunodeficiency conferred by the disease and therapeutics.6 The immunogenicity of the COVID-19 vaccines in patients with exposure to CD19-directed chimeric antigen receptor (CAR) T-cell therapy is not established. CD19 CAR T-cell therapy results in B-cell aplasia, which in turn can affect humoral and cellular immune responses against novel antigens and vaccination.7 We present results from our clinical study to evaluate immune responses against messenger RNA (mRNA)–based COVID-19 vaccines in patients with lymphoma who have received CD19-directed CAR T-cell therapy.
All patients and healthy controls (HCs) were enrolled in a prospective clinical study evaluating immune responses to COVID-19 mRNA vaccines authorized by the US Food and Drug Administration in the United States. Plasma samples were generated from heparinized peripheral blood of 4 HCs receiving the same vaccines and 18 patients with B-cell lymphoma treated with CD19 CAR T cells (supplemental Table 1). Samples collected at 4 weeks after the second dose of the vaccine (day 56) were available for 14 patients treated with CAR T cells, and 4 samples from patients treated with CAR T cells were available from 4 weeks after the first dose (day 28). Median follow-up period after the first dose of vaccine was 129 days (range, 60-155). Median age of the patients with lymphoma was 50.5 years (range, 24-87). Ten patients had large B-cell lymphoma, 5 had follicular lymphoma, and 3 had mantle cell lymphoma. Sixteen patients had advanced-stage (III/IV) disease and had received a median of 3 prior lines of therapy (range, 2-7). All but 2 patients were in complete remission at the time of vaccination (supplemental Figure 1). Ten patients received the mRNA-1273 (Moderna) vaccine, and 8 received the BNT162b2 (Pfizer/BioNTech) vaccine. All 18 patients had received CD19-directed CAR T-cell therapy for relapsed or refractory B-cell malignancies. Two patients received the vaccine before CAR T-cell therapy (second dose 48 and 97 days before CAR T cells, respectively), 13 patients received the vaccine after CAR T-cell therapy (median, 33 days after CAR T cells; range, 24-447), and 3 patients were treated with CAR T-cell therapy followed by allogeneic stem cell transplantation (alloSCT) and then received the vaccine (median, 499 days after CAR T cells; range, 466-532; supplemental Figure 1).
Although the peripheral blood plasma from 3 of 4 HCs already showed substantial SARS-CoV-2–neutralizing activity, with 4 of 4 showing some neutralizing activity at ∼4 weeks after the first dose of the COVID-19 mRNA vaccine, none of the 4 patients treated with CD19-directed CAR T-cell therapy showed any antibody-mediated neutralizing activity in their blood at the same point in time (Figure 1A). Upon further follow-up, these 4 patients had no neutralizing activity at day 84. At ∼4 weeks after receiving the second dose of the vaccine, all 4 HCs had complete or almost complete neutralizing activity (Figure 1B). In marked contrast, only 1 of 14 patients treated with CAR T-cell therapy showed any relevant antibody-mediated SARS-CoV-2–neutralizing activity in their blood (Figure 1B).
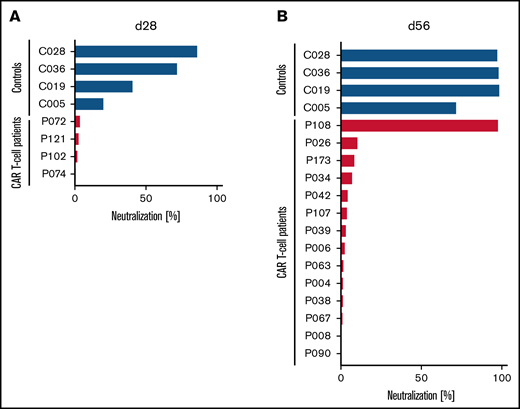
Lack of SARS-CoV-2–neutralizing activity in patients treated with CD19-directed CAR T-cell therapy receiving mRNA vaccines. Neutralizing activity of vaccine-induced anti-RBD antibodies in the peripheral blood of patients treated with CD19-directed CAR T-cell therapy (red bars) and HCs (blue bars) after the first (A) or second (B) dose of the vaccine was measured as the degree of inhibition of interactions between RBD and angiotensin-converting enzyme-2 (ACE2). Additionally, for patients P072, P121, P102, and P074, day-84 samples showed complete lack of neutralizing antibodies. Of note, P108 (only patient with positive neutralization assay) had no detectable circulating B cells, suggesting the possibility of antibody production from nodal B cells or plasma cells. Neutralizing activity was measured using the cPass Neutralization Antibody Detection Kit (GenScript Biotech), which is a surrogate test detecting circulating neutralizing antibodies against SARS-CoV-2 that block the interaction between the RBD of the viral spike glycoprotein and the ACE2 cell-surface receptor.
Lack of SARS-CoV-2–neutralizing activity in patients treated with CD19-directed CAR T-cell therapy receiving mRNA vaccines. Neutralizing activity of vaccine-induced anti-RBD antibodies in the peripheral blood of patients treated with CD19-directed CAR T-cell therapy (red bars) and HCs (blue bars) after the first (A) or second (B) dose of the vaccine was measured as the degree of inhibition of interactions between RBD and angiotensin-converting enzyme-2 (ACE2). Additionally, for patients P072, P121, P102, and P074, day-84 samples showed complete lack of neutralizing antibodies. Of note, P108 (only patient with positive neutralization assay) had no detectable circulating B cells, suggesting the possibility of antibody production from nodal B cells or plasma cells. Neutralizing activity was measured using the cPass Neutralization Antibody Detection Kit (GenScript Biotech), which is a surrogate test detecting circulating neutralizing antibodies against SARS-CoV-2 that block the interaction between the RBD of the viral spike glycoprotein and the ACE2 cell-surface receptor.
Two patients who received the vaccine first and then CAR T-cell therapy (with no other therapy or IV immunoglobulin in between) had no neutralizing antibodies at the day-56 assessment postvaccination. Twelve patients received CAR T-cell therapy and then the vaccine; with the exception of 1 patient, none of these patients had neutralizing antibodies. Of note, none of the 12 patients received any maintenance or salvage therapy or IV immunoglobulin before assessment of vaccine immune response. Three patients who received CAR T-cell therapy and then underwent alloSCT, followed by vaccination, also did not have any neutralizing antibodies. Although alloSCT may have contributed to poor immunogenicity, prior CD19-directed CAR T-cell therapy and potential persistence of CAR T cells, especially after reduced-intensity conditioning, may have affected humoral response in these patients through long-term depletion of both recipient and donor B cells.
Upon assessing whether globally insufficient antibody-mediated immunity was the underlying cause of the lack of response to the COVID-19 vaccine in our patients treated with CAR T-cell therapy, we found that these patients indeed showed lower levels of total immunoglobulin G (IgG), IgM, and IgA compared with controls (Figure 2A). However, IgG antibody levels against common microbial and viral antigens, including vaccine-induced immune responses against influenza and tetanus toxoid, were comparable to those observed in HCs (Figure 2B). In marked contrast, although at 4 weeks after the second dose of the vaccine the HCs showed high levels of vaccine-induced IgG antibody titers against all the viral spike proteins (S1, S2, RBD), including the delta variants of the S1 and RBD proteins (Figure 2C), this was not the case for our patients treated with CAR T-cell therapy. A vast majority of our patients treated with CAR T-cell therapy did not show IgG antibody responses against any of the SARS-CoV-2 proteins analyzed (Figure 2C). Importantly, there was a strong correlation between IgG antibody titers against the RBD protein and the level of SARS-CoV-2–neutralizing activity (Figure 2D), highlighting the potential clinical relevance of our serological findings. Because it has previously been shown that the mRNA vaccines are capable of inducing IgA antibody responses,8 we also measured antiviral IgA in our patients, and we found that, similarly to IgG anti–SARS-CoV-2 immune responses, the controls showed IgA antibodies against all viral proteins except the N protein, but patients receiving CAR T-cell therapy did not show any antigen-specific IgA antibodies (Figure 2E).
![Patients treated with CD19-directed CAR T-cell therapy receiving mRNA vaccines are incapable of generating new anti–SARS-CoV-2 IgG and IgA antibodies. (A) Absolute levels of IgG, IgM, and IgA antibodies in our study participants were measured using a commercially available enzyme-linked immunosorbent assay (ELISA; Invitrogen, Waltham, MA). (B) IgG antibodies against full-length recombinant influenza A nucleoprotein (Flu), tetanus toxoid (TT), Epstein-Barr virus glycoprotein gp350 (EBV), cytomegalovirus (CMV), and herpes simplex virus type 1 (HSV) glycoprotein D were measured in an ELISA as positive controls. IgG (C) and IgA (E) antibody titers against different SARS-CoV-2 S proteins, including their delta variants (RBD [L452R, T478K] and S1 [T19R, G142D, EF156-157del, R158G, L452R, T478K, D614G, P681R]), and SARS-CoV-2 N proteins were also measured in an ELISA. Values calculated represent reciprocal end point titers, and groups were compared using the Mann-Whitney U test. (D) Correlations between anti-RBD IgG antibody titers and neutralizing activity in the same patient or control. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/6/2/10.1182_bloodadvances.2021006112/5/m_advancesadv2021006112f2.png?Expires=1769145488&Signature=vHCKKpVXL9XmBrspqDtZz4g58A-BSklSNl~6lhJjham9P7IuzmKSi3Wl7j9VhA8SrSA5whanmqda~CRUtQq0gNYyLrgh-cqlTo3NRxpNT6AVs5uitE03cKiHqNbzMMEvnEmJmh9shkZ1LCZcS4RsKIew-Y1EzspGZ35UuJUzkiGc8zC9xMV2uKWk2OS67xImXPRwHKFhZtmVQrnhutYdCVW7-1V6SU1D~-rRfVpATpUHPLvCyu9FA9ah1shjx0i4-CMiEnig6QrIK-1qV~NsXtXjffAzcLqKMFF2MEORDNdbpTYlKaAGIbxuZVmqj2OFmt0Rxz4nrvXxJxTHRqoceQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Patients treated with CD19-directed CAR T-cell therapy receiving mRNA vaccines are incapable of generating new anti–SARS-CoV-2 IgG and IgA antibodies. (A) Absolute levels of IgG, IgM, and IgA antibodies in our study participants were measured using a commercially available enzyme-linked immunosorbent assay (ELISA; Invitrogen, Waltham, MA). (B) IgG antibodies against full-length recombinant influenza A nucleoprotein (Flu), tetanus toxoid (TT), Epstein-Barr virus glycoprotein gp350 (EBV), cytomegalovirus (CMV), and herpes simplex virus type 1 (HSV) glycoprotein D were measured in an ELISA as positive controls. IgG (C) and IgA (E) antibody titers against different SARS-CoV-2 S proteins, including their delta variants (RBD [L452R, T478K] and S1 [T19R, G142D, EF156-157del, R158G, L452R, T478K, D614G, P681R]), and SARS-CoV-2 N proteins were also measured in an ELISA. Values calculated represent reciprocal end point titers, and groups were compared using the Mann-Whitney U test. (D) Correlations between anti-RBD IgG antibody titers and neutralizing activity in the same patient or control. ns, not significant.
Patients treated with CD19-directed CAR T-cell therapy receiving mRNA vaccines are incapable of generating new anti–SARS-CoV-2 IgG and IgA antibodies. (A) Absolute levels of IgG, IgM, and IgA antibodies in our study participants were measured using a commercially available enzyme-linked immunosorbent assay (ELISA; Invitrogen, Waltham, MA). (B) IgG antibodies against full-length recombinant influenza A nucleoprotein (Flu), tetanus toxoid (TT), Epstein-Barr virus glycoprotein gp350 (EBV), cytomegalovirus (CMV), and herpes simplex virus type 1 (HSV) glycoprotein D were measured in an ELISA as positive controls. IgG (C) and IgA (E) antibody titers against different SARS-CoV-2 S proteins, including their delta variants (RBD [L452R, T478K] and S1 [T19R, G142D, EF156-157del, R158G, L452R, T478K, D614G, P681R]), and SARS-CoV-2 N proteins were also measured in an ELISA. Values calculated represent reciprocal end point titers, and groups were compared using the Mann-Whitney U test. (D) Correlations between anti-RBD IgG antibody titers and neutralizing activity in the same patient or control. ns, not significant.
Six patients enrolled in this study had received a third dose of the same COVID-19 vaccine at the time of this report. Neutralization assay and antibody levels were negative in all 6 patients ∼1 month after the third dose (median, 37 days; supplemental Table 2).
As of the last follow-up, none of these patients reported symptoms or tested positive for COVID-19. This may be a result of the relatively low infection rate in this part of the country before the data cutoff and strong recommendations for enhanced infection precautions, like avoiding crowds and masking. This finding may also be a result of immune protection mediated by T-cell response, which was not analyzed in these patients at the time of this report.9
Studies have shown ongoing CD20-directed therapy and exposure may affect humoral response in patients with B-cell malignancies.10 Although the impact of prior CD20 therapy cannot be fully assessed, given that the median time from exposure to CD20 therapy in our study was 44 months (range, 3-311) and 95% of patients did not exhibit neutralization (a rate too high to be attributed to CD20 therapy alone), it is quite likely that the poor immunogenicity seen in this study was chiefly a result of CD19-directed CAR T-cell therapy. Examination of the role of T cells in conferring protective immunity and assessment of the impact of a third dose of an mRNA vaccine in all of these patients is underway.
In conclusion, in this prospectively conducted clinical study, 17 of 18 patients with lymphoma who received CD19-directed CAR T-cell therapy had very poor immunoreactivity against mRNA-based COVID-19 vaccines as measured by neutralization assays and antibody titers. Additional studies are needed to demonstrate that seronegativity in this patient population indeed results in higher incidence of breakthrough infections. The antibody titers against B.1.617.2 (delta variant, S1, and RBD protein) were also demonstrably poor. Importantly, the antibody response to common pathogens (eg, influenza, Epstein-Barr virus, and tetanus toxoid) was preserved, suggesting impaired immune response primarily against novel antigens like SARS-COV-2.
Acknowledgments: Supported by University of Maryland Greenebaum Cancer Center grant P30 CA134274 from the National Cancer Institute, National Institutes of Health, and a grant from the Kahlert Family Foundation.
Contribution: S.D., A.P.R., and D.A. were responsible for study conceptualization and design; all authors were responsible for provision of study materials or patients; T.L., S.A., T.I., and D.A. performed immunological analyses; and S.D., F.L., A.P.R., and D.A. were responsible for collection and assembly of data. All authors performed data analysis and interpretation, contributed to manuscript writing, and provided final approval of the manuscript.
Conflict-of-interest disclosure: S.D. serves on advisory boards for Bristol-Myers Squibb, Incyte, and Atara Biotherapeutics. The remaining authors declare no competing financial interests.
Correspondence: Saurabh Dahiya, Greenebaum Comprehensive Cancer Center, University of Maryland, 22 S. Greene St, N9E12, Baltimore, MD 21201; e-mail: sdahiya@umm.edu.

