

Key Points
Concizumab efficacy was maintained during the extension parts of phase 2 trials.
Treatment was well tolerated across hemophilia subtypes for a period of at least 76 weeks.

Abstract
Despite current therapies, there remains an unmet need for treatment for patients with hemophilia. The main parts of two phase 2 trials established clinical proof-of-concept for once-daily, subcutaneous concizumab prophylaxis in patients with hemophilia A/B with inhibitors (HAwI/HBwI; explorer4) and severe hemophilia A without inhibitors (HA; explorer5). Here, we present results from extension parts of these trials, included to evaluate longer term safety and efficacy. Both trials included main (≥24 weeks) and extension (52-102 weeks) parts, with patients receiving concizumab 0.15 mg/kg with potential dose escalation to concizumab 0.20 or 0.25 mg/kg if they experienced ≥3 treated spontaneous bleeding episodes within 12 weeks. Endpoints included annualized bleeding rate (ABR), adverse events (AEs), and occurrence of antidrug antibodies. Thromboembolic events were AEs of special interest. Thirty-six patients with HA, 15 with HAwI, and 10 with HBwI were exposed to concizumab. Estimated ABRs during the main + extension parts at last dose level were 4.8 (95% confidence interval [CI], 3.2-7.2) and 6.4 (95% CI, 4.1-9.9) in explorer4 and explorer5, respectively (spontaneous ABRs were 1.8 [95% CI, 1.2-2.6] and 2.1 [95% CI, 1.3-3.3]). Most AEs were mild, with no deaths, events leading to withdrawal, or thromboembolic events. Anti-drug antibodies developed in 25% of patients and were low titer and transient, with no observed clinical effect in most cases. Results of the main + extension parts of these trials were consistent with results of the main parts. Ongoing phase 3 trials will further evaluate concizumab as a once-daily, subcutaneous treatment across hemophilia subtypes. These trials were registered at www.clinicaltrials.gov as #NCT03196284 and #NCT03196297.
Introduction
Current guidelines for the treatment of patients with hemophilia recommend replacement therapy with the coagulation factor that is deficient (factor VIII [FVIII] in hemophilia A [HA] and factor IX [FIX] in hemophilia B [HB]).1,2 However, the need for regular intravenous infusions is a drawback of factor replacement therapy and represents a significant treatment burden that may affect patient adherence.3 In addition, the efficacy of factor replacement therapy may be suboptimal in some cases and can be associated with the development of neutralizing antibodies (inhibitors), which remains a major limitation of hemophilia therapy.4 Approximately 25% to 30% of patients with severe HA2,5,6 and 1% to 10% with severe HB7-9 develop inhibitors, which can result in complete or partial inactivation of the infused replacement factor, leading to an increased burden of disease for these patients.2,5 Novel treatment options are particularly needed for patients with HB with inhibitors (HBwI): anaphylactic or severe allergic reactions to infused FIX occur in ∼50% of these patients, and many exhibit a poor clinical response to immune tolerance induction (a process of repeated regular dosing of FIX that aims to induce tolerance in patients with inhibitors).2,10,11
Due to the ongoing unmet needs of patients with hemophilia, recent research has focused on development of non-factor replacement therapies. One alternative is the use of non-factor therapies to enhance coagulation or target anticoagulation pathways.12,13 Several monoclonal antibodies targeting coagulation pathways have been investigated as treatment options for patients with hemophilia, including emicizumab, a bispecific antibody for the treatment of HA (with and without inhibitors) targeting activated factor IX (FIXa)/FIX and FX/activated factor X (FXa),13 and BAY1093884, a monoclonal antibody targeting tissue factor pathway inhibitor (TFPI).12 Although results have been encouraging with some of these treatments, an important safety consideration is the occurrence of thromboembolic events (TEs), with the development of BAY1093884 terminated in 2019 after the occurrence of 3 TEs in 24 patients during a phase 2 trial.12,14
Concizumab is an anti-TFPI antibody for subcutaneous prophylaxis across all hemophilia subtypes that acts independently from FVIII and FIX by enhancing the initiation phase of coagulation through increased FXa production, allowing sufficient thrombin generation (TG) to prevent bleeds.15 The pharmacodynamic relationship between concizumab exposure, free TFPI, and TG was previously confirmed in a phase 1b trial.16 Two phase 2 trials were initiated to evaluate the efficacy and safety of subcutaneous concizumab prophylaxis in patients with HA with inhibitors (HAwI)/HBwI (explorer4) and severe hemophilia A without inhibitors (HA; explorer5). Results from the main part (at least 24 weeks of treatment) of both trials showed that concizumab was well tolerated, with the potential to provide prophylactic bleed protection regardless of hemophilia subtype.17 Here, we report the results from the main + extension parts of these studies, which were included to assess the longer term efficacy and safety of concizumab.
Methods
Trial design
Full details of the study design of explorer4 (clinicaltrials.gov identifier #NCT03196284; inhibitor trial) and explorer5 (clinicaltrials.gov identifier #NCT03196297; non-inhibitor trial) have previously been reported by Shapiro et al.17 Briefly, both trials comprised a main part (at least 24 weeks) in which clinical proof-of-concept was established, and an extension part (52-102 weeks) to evaluate longer term efficacy and safety of concizumab (giving a total of ≥76 weeks of treatment). explorer4 was initiated on 10 August 2017 and completed on 31 January 2020, and explorer5 began on 16 August 2017 and was completed on 3 June 2020.
explorer4 was an open-label, multicenter, randomized controlled trial conducted across 17 sites in 12 countries. Patients were randomized 2:1 to receive prophylaxis with concizumab or on-demand treatment with eptacog alfa activated (recombinant activated factor VII [rFVIIa]; NovoSeven® [Novo Nordisk], dosed at the investigator’s discretion) during the main part of the trial. All patients who received rFVIIa during the main part were switched to daily concizumab treatment during the extension part (≥52 weeks). During the open-label, multicenter, single-arm explorer5 trial (conducted across 26 sites in 11 countries), all patients received prophylactic treatment with concizumab during the main and extension parts.
The dosing regimen for both phase 2 studies was selected based on pharmacokinetic/pharmacodynamic modeling of results from phase 1 studies (further details regarding this selection have been reported previously).17,18 Patients received daily subcutaneous injections of concizumab 0.15 mg/kg, with potential dose escalation to concizumab 0.20 or 0.25 mg/kg (Figure 1). In the explorer4 trial, an initial loading dose of 0.5 mg/kg concizumab was administered, whereas patients enrolled in explorer5 did not receive a loading dose. After 1 week of concizumab dosing (0.15 mg/kg) in explorer4, a dose of rFVIIa 90 µg/kg was administered to patients in a nonbleeding state to assess the safety of coadministration of concizumab and rFVIIa.
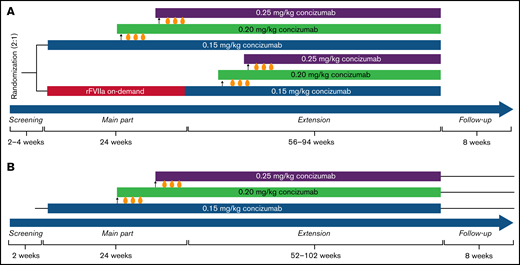
Study design for the phase 2 concizumab trials. (A) explorer4 (HAwI/HBwI). (B) explorer5 (HA). As shown in panel A, patients were randomized 2:1 to receive either prophylaxis with concizumab or on-demand treatment with rFVIIa for 24 weeks (main part). Patients who received rFVIIa during the main part were switched to concizumab prophylaxis during the extension part (56-94 weeks). As shown in panel B, all patients received concizumab prophylaxis during the main part (24 weeks) and extension part (52-102 weeks). Dose escalation criteria throughout both trials were ≥3 treatment-requiring spontaneous bleeding episodes within the 12 weeks before concizumab treatment during both the main and extension parts.
Study design for the phase 2 concizumab trials. (A) explorer4 (HAwI/HBwI). (B) explorer5 (HA). As shown in panel A, patients were randomized 2:1 to receive either prophylaxis with concizumab or on-demand treatment with rFVIIa for 24 weeks (main part). Patients who received rFVIIa during the main part were switched to concizumab prophylaxis during the extension part (56-94 weeks). As shown in panel B, all patients received concizumab prophylaxis during the main part (24 weeks) and extension part (52-102 weeks). Dose escalation criteria throughout both trials were ≥3 treatment-requiring spontaneous bleeding episodes within the 12 weeks before concizumab treatment during both the main and extension parts.
Dose escalation criteria were ≥3 treatment-requiring spontaneous bleeding episodes within 12 weeks while receiving concizumab treatment during the main and extension parts of both trials. Breakthrough bleeding episodes requiring treatment were managed with rFVIIa in explorer4 (provided there were no safety concerns after the dose, delivered in a nonbleeding state as described earlier) and with nonmodified FVIII in explorer5 (either Novoeight®, provided by Novo Nordisk, or another nonmodified FVIII product that was not provided by Novo Nordisk).
The explorer4 and explorer5 trials were conducted in accordance with the Declaration of Helsinki and International Conference on Harmonisation Good Clinical Practice Guideline. Patients provided written informed consent, and both trials were approved by an independent ethics committee or institutional review board as required according to local regulations. In addition, an independent external data monitoring committee was established to review the accumulating data at regular intervals to protect the safety of the patients. All authors had access to the clinical trial data, which were analyzed by Novo Nordisk.
Temporary clinical pause of concizumab treatment during explorer5
On 16 March 2020, Novo Nordisk informed regulatory authorities and investigators of the company’s decision to pause concizumab treatment in ongoing clinical trials, which included explorer5 at the time, due to reports of 5 nonfatal thrombotic serious adverse events (AEs) in 3 patients enrolled in the phase 3 trials explorer7 (patients with HAwI/HBwI; clinicaltrials.gov identifier #NCT04083781) and explorer8 (patients with HA/HB; clinicaltrials.gov identifier #NCT04082429) while receiving concizumab treatment (under a schedule of administration different from that of explorer4 and explorer5). These AEs were acute myocardial infarction in 1 patient with HA, a renal infarction in 1 patient with HBwI, and 3 TEs (deep venous thrombosis, pulmonary embolism, and superficial thrombosis of vein [left elbow region at site of FVIII injection]) in 1 patient with HA.19
Around the same time, the COVID-19 outbreak was declared a global pandemic by the World Health Organization. Treatment with concizumab was stopped in the remaining patients in explorer5 by 18 March 2020. The explorer4 trial had already been completed on 31 January 2020. Overall, the concizumab pause and the COVID-19 pandemic did not affect the efficacy and safety endpoints in the explorer5 trial as all patients had already completed at least 76 weeks of treatment with concizumab (the predefined time frame for key endpoints).
Trial population
The study population of explorer4 comprised male patients with HAwI/HBwI aged ≥18 years with a documented history of high-titer (≥5 Bethesda units) inhibitors, and the explorer5 study included male patients aged ≥18 years with severe HA (defined by FVIII activity <1%). Patients receiving on-demand treatment were eligible if they had experienced ≥6 bleeding episodes in the 24 weeks before screening (or 12 bleeding episodes during 52 weeks).
Exclusion criteria for both trials included diagnosis with a bleeding disorder other than hemophilia, known or suspected hypersensitivity to trial products or related products, major surgery within 1 month before initiation of trial activities or planned surgical procedures during the trial, history or high risk of thromboembolic disease, and systemic inflammatory condition requiring systemic treatment. In addition, patients with ongoing or planned immune tolerance induction therapy with FVIII or FIX were excluded from explorer4. Further details of these criteria have been reported previously by Shapiro et al.17
Objectives, endpoints, and assessments
The explorer4 and explorer5 trials were designed to assess the efficacy and safety of once-daily subcutaneous concizumab in preventing bleeding episodes in patients with HAwI and HBwI (explorer4) and patients with severe HA (explorer5), with extension parts included to evaluate the longer term efficacy and safety of treatment over a period of at least 76 weeks. The protocol was amended to prolong the duration of the extension parts as needed to allow patients to continue treatment in subsequent phase 3 trials. In addition, both trials assessed the immunogenicity of concizumab and patient-reported outcomes.
The primary endpoint was number of treated bleeding episodes during at least 24 weeks from treatment initiation (main part) and has previously been described by Shapiro et al.17 Bleeding episodes were evaluated according to World Federation of Hemophilia guidance (based on cause [spontaneous, traumatic], location, and severity).20 Secondary efficacy endpoints evaluated during the main + extension parts included number of treated bleeding episodes and number of spontaneous bleeding episodes during at least 76 weeks of treatment. Secondary pharmacokinetic and pharmacodynamic endpoints included concentration of concizumab, free TFPI concentration, peak TG, and TG potential, all before last dose administration at 76 weeks.
Secondary safety endpoints for both trials included number of treatment-emergent AEs; occurrence of anti-drug antibodies (ADAs); and change from baseline of D-dimer concentrations, prothrombin fragment 1 + 2, and other coagulation parameters (including fibrinogen, prothrombin time, activated partial thromboplastin time, and antithrombin) during at least 76 weeks of treatment. ADA assessment was conducted according to international guidelines and recommendations using a bridging electrochemiluminescence assay, with labeled concizumab used for antibody detection (assay sensitivity was 1 ng/mL). Positive samples were additionally analyzed for neutralizing activity using a modified chromogenic assay (full details previously described by Shapiro et al17 ). Additional safety evaluations included physical examination and vital sign measurements. TEs were considered as AEs of special interest, and AEs requiring additional data collection included injection site reactions, hypersensitivity type reactions (including anaphylactic reactions), and medication errors.
Statistical analysis
The full analysis and safety analysis sets for both trials comprised all patients dosed with concizumab. Data were analyzed by using SAS version 9.4 (SAS Institute, Inc.), and efficacy results for the main + extension parts of the trials are presented for the last concizumab dose level reached; that is, the individual last dose level (previous dose levels disregarded), with the corresponding results for annualized bleeding rates (ABRs) during the entire exposure period reported in the supplemental Materials.
Details of the statistical analysis of the primary endpoint of both trials have been reported previously.17 The supportive secondary efficacy endpoints of number of bleeding episodes and number of spontaneous bleeding episodes (during at least 76 weeks from treatment onset) were estimated by using the same negative binomial regression model as the primary endpoint (with log of exposure time as offset) and by performing the same analyses as for the primary endpoint; one included only observations from the period on the last dose level and one included the entire escalation pattern. ABR according to hemophilia type was estimated as part of a post hoc analysis based on a negative binomial regression with log of exposure time as offset and hemophilia type as factor.
An additional post hoc analysis was performed to estimate the ABR in patients who received on-demand treatment with rFVIIa during the main part and concizumab treatment during the extension part of explorer4, using a negative binomial regression with log of exposure time as offset and treatment regimen as factor. The within-subject correlation was estimated by a generalized estimating equation analysis, with a working independence covariate structure and using a robust estimator.
Results
Trial population and baseline characteristics
In the explorer4 trial, 26 patients were randomized in the main part, with 17 patients receiving concizumab treatment and 9 patients treated with rFVIIa on-demand (1 patient discontinued before the end of the main part and did not enter the extension). The 25 patients who completed the main part and went on to enter the extension part comprised the full analysis and safety analysis sets (15 patients with HAwI and 10 patients with HBwI) (Figure 2). Twenty-two patients completed the extension part, with one withdrawing due to protocol-defined lack of efficacy on the last dose level, one due to suspicion of no therapeutic effect due to normal TFPI level with presence of ADAs (as discussed in the Immunogenicity section), and one after withdrawal of consent.
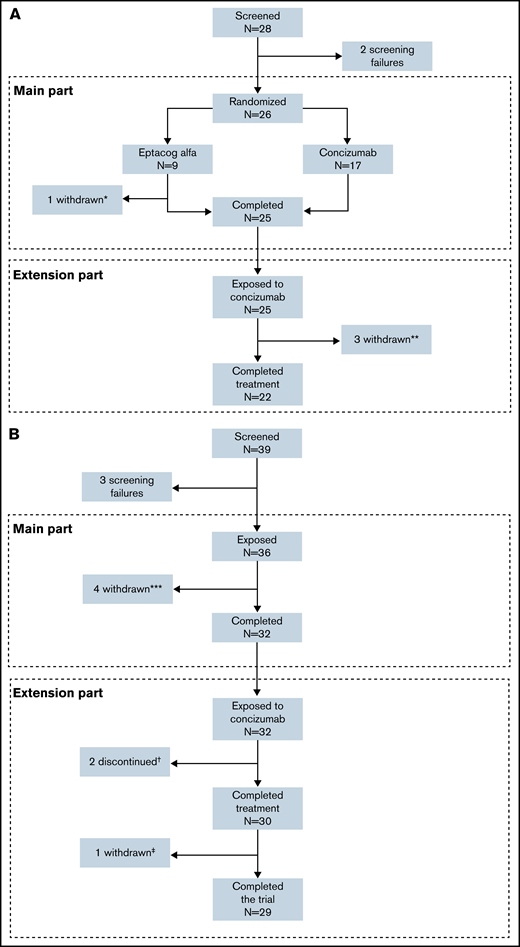
Patient disposition in the phase 2 concizumab trials. (A) explorer4 (HAwI, HBwI). (B) explorer5 (HA). *One patient withdrew consent after randomization. **Three patients withdrew in the extension phase (one due to lack of efficacy, one because of suspicion of no therapeutic effect due to normal TFPI level with ADA, and one withdrawal of consent). ***Four patients withdrew before the end of the main part. †Two patients discontinued due to lack of efficacy. ‡One patient withdrew consent in the extension part.
Patient disposition in the phase 2 concizumab trials. (A) explorer4 (HAwI, HBwI). (B) explorer5 (HA). *One patient withdrew consent after randomization. **Three patients withdrew in the extension phase (one due to lack of efficacy, one because of suspicion of no therapeutic effect due to normal TFPI level with ADA, and one withdrawal of consent). ***Four patients withdrew before the end of the main part. †Two patients discontinued due to lack of efficacy. ‡One patient withdrew consent in the extension part.
Thirty-two patients with HA completed the main part of explorer5 and went on to enter the extension (4 patients withdrew during the main part), 29 of whom completed both parts. The full analysis and safety analysis sets included all 36 patients who were exposed to concizumab during the trial. One patient who completed treatment withdrew from the trial before the follow-up visit with no stated reason, and 2 patients withdrew due to protocol-defined lack of efficacy at last dose level.
Patient characteristics at baseline were similar in both trials, and full details have been described previously.17 In brief, mean patient age was 35.4 years in the explorer4 trial and 36.9 years in explorer5, and mean time since diagnosis was 34.7 and 34.4 years in explorer4 and explorer5, respectively.
Temporary clinical pause of concizumab treatment and impact of COVID-19 on explorer5
When a pause of concizumab treatment during ongoing clinical trials was implemented on 16 March 2020, thirteen patients enrolled in explorer5 were still receiving treatment. These patients were requested to discontinue treatment with concizumab and attend the end-of-treatment visit (Visit 16) as soon as possible (as per protocol), with the last patient receiving a concizumab dose on 18 March 2020. Further details of the impact of the COVID-19 pandemic on the explorer5 trial are provided in the supplemental Materials.
Longer term efficacy in the explorer4 and explorer5 trials
Concizumab dose escalation and exposure during the extension parts.
During the extension part of explorer4, twelve of 25 patients remained on a dose of concizumab 0.15 mg/kg (7 patients with HAwI and 5 patients with HBwI) (Table 1). The concizumab dose was escalated to 0.20 mg/kg for 6 patients with HAwI and 3 patients with HBwI, and 2 patients with HAwI and 2 with HBwI were escalated to concizumab 0.25 mg/kg during the extension part. In the explorer5 trial, 15 patients remained on their original dose of concizumab 0.15 mg/kg, with 10 and 11 patients escalating to concizumab 0.20 mg/kg and 0.25 mg/kg, respectively. Details of patients with a dose escalation during the main parts of both trials have been reported previously.17 The total cumulative concizumab exposure time, including follow-up for patients during the main + extension parts, was 71.9, 27.0, and 18.3 years for patients with HA, HAwI, and HBwI (Table 2).
Concizumab dose escalation during the main + extension parts of explorer4 (HAwI, HBwI) and explorer5 (HA) according to hemophilia subtype; full analysis set
| Dose escalation* | HA (n = 36) | HAwI (n = 15) | HBwI (n = 10) |
|---|---|---|---|
| None, 0.15 mg /kg only | 15 (41.7) | 7 (46.7) | 5 (50.0) |
| One, 0.15 and 0.20 mg/kg | 10 (27.8) | 6 (40.0) | 3 (30.0) |
| Two, 0.15, 0.20, and 0.25 mg/kg | 11 (30.6)† | 2 (13.3) | 2 (20.0) |
| Dose escalation* | HA (n = 36) | HAwI (n = 15) | HBwI (n = 10) |
|---|---|---|---|
| None, 0.15 mg /kg only | 15 (41.7) | 7 (46.7) | 5 (50.0) |
| One, 0.15 and 0.20 mg/kg | 10 (27.8) | 6 (40.0) | 3 (30.0) |
| Two, 0.15, 0.20, and 0.25 mg/kg | 11 (30.6)† | 2 (13.3) | 2 (20.0) |
Data are presented as n (percentage of patients) with dose escalation.
One HA patient with 2 escalations was later de-escalated to concizumab 0.15 mg/kg, and one HA patient with 2 escalations was later de-escalated to concizumab 0.20 mg/kg before a further escalation to concizumab 0.25 mg/kg.
Treatment-emergent AEs according to hemophilia subtype during the main + extension parts of explorer4 (HAwI, HBwI) and explorer5 (HA); safety analysis set
| Hemophilia subtype | HA (n = 36) | HAwI (n = 15) | HBwI (n = 10) | |||
|---|---|---|---|---|---|---|
| Cumulative concizumab exposure time, including follow-up, y | 71.9 | 27.0 | 18.3 | |||
| Hemophilia subtype | HA (n = 36) | HAwI (n = 15) | HBwI (n = 10) | |||
|---|---|---|---|---|---|---|
| Cumulative concizumab exposure time, including follow-up, y | 71.9 | 27.0 | 18.3 | |||
| n (%) | E [R] | n (%) | E [R] | n (%) | E [R] | |
|---|---|---|---|---|---|---|
| All AEs | 33 (91.7) | 298 [4.1] | 13 (86.7) | 67 [2.5] | 9 (90.0) | 64 [3.5] |
| Serious AEs | 5 (13.9) | 5 [0.1] | 0 | 5 (50.0) | 9 [0.5] | |
| Severe | 4 (11.1) | 4 [0.1] | 0 | 2 (20.0) | 4 [0.2] | |
| Fatal | 0 | 0 | 0 | |||
| AEs leading to withdrawal | 0 | 0 | 0 | |||
| AESI (thromboembolic events) | 0 | 0 | 0 | |||
| Most frequent AEs by PT and SOC, ≥5% | ||||||
| Gastrointestinal disorders | ||||||
| Dental caries | 3 (8.3) | 6 [0.1] | 0 | 1 (10.0) | 2 [0.1] | |
| Diarrhea | 1 (2.8) | 1 [0.0] | 3 (20.0) | 3 [0.1] | 1 (10.0) | 2 [0.1] |
| General disorders and administration site conditions | ||||||
| Injection site reactions* | 14 (38.9) | 25 [0.3] | 2 (13.3) | 3 [0.1] | 4 (40.0) | 15 [0.8] |
| Pyrexia | 3 (8.3) | 4 [0.1] | 2 (13.3) | 2 [0.1] | 0 | |
| Infections and infestations | ||||||
| Gastrointestinal infection | 3 (8.3) | 3 [0.0] | 1 (6.7) | 1 [0.0] | 0 | |
| Influenza | 4 (11.1) | 4 [0.1] | 1 (6.7) | 1 [0.0] | 0 | |
| Nasopharyngitis | 12 (33.3) | 22 [0.3] | 0 | 3 (30.0) | 6 [0.3] | |
| Pharyngitis | 3 (8.3) | 3 [0.0] | 1 (6.7) | 2 [0.1] | 0 | |
| Rhinitis | 2 (5.6) | 2 [0.0] | 2 (13.3) | 2 [0.1] | 0 | |
| Upper respiratory tract infection | 5 (13.9) | 7 [0.1] | 2 (13.3) | 4 [0.1] | 2 (20.0) | 4 [0.2] |
| Investigations | ||||||
| D-dimer level increased | 9 (25.0) | 12 [0.2] | 1 (6.7) | 1 [0.0] | 1 (10.0) | 1 [0.1] |
| Prothrombin fragments 1 + 2 increased | 7 (19.4) | 12 [0.2] | 2 (13.3) | 2 [0.1] | 0 | |
| Musculoskeletal and connective tissue disorders | ||||||
| Arthralgia | 4 (11.1) | 6 [0.1] | 3 (20.0) | 3 [0.1] | 0 | |
| Back pain | 5 (13.9) | 7 [0.1] | 0 | 0 | ||
| Nervous system disorders | ||||||
| Headache | 8 (22.2) | 12 [0.2] | 1 (6.7) | 1 [0.0] | 1 (10.0) | 1 [0.1] |
| Respiratory, thoracic, and mediastinal disorders | ||||||
| Cough | 4 (11.1) | 4 [0.1] | 0 | 0 | ||
| n (%) | E [R] | n (%) | E [R] | n (%) | E [R] | |
|---|---|---|---|---|---|---|
| All AEs | 33 (91.7) | 298 [4.1] | 13 (86.7) | 67 [2.5] | 9 (90.0) | 64 [3.5] |
| Serious AEs | 5 (13.9) | 5 [0.1] | 0 | 5 (50.0) | 9 [0.5] | |
| Severe | 4 (11.1) | 4 [0.1] | 0 | 2 (20.0) | 4 [0.2] | |
| Fatal | 0 | 0 | 0 | |||
| AEs leading to withdrawal | 0 | 0 | 0 | |||
| AESI (thromboembolic events) | 0 | 0 | 0 | |||
| Most frequent AEs by PT and SOC, ≥5% | ||||||
| Gastrointestinal disorders | ||||||
| Dental caries | 3 (8.3) | 6 [0.1] | 0 | 1 (10.0) | 2 [0.1] | |
| Diarrhea | 1 (2.8) | 1 [0.0] | 3 (20.0) | 3 [0.1] | 1 (10.0) | 2 [0.1] |
| General disorders and administration site conditions | ||||||
| Injection site reactions* | 14 (38.9) | 25 [0.3] | 2 (13.3) | 3 [0.1] | 4 (40.0) | 15 [0.8] |
| Pyrexia | 3 (8.3) | 4 [0.1] | 2 (13.3) | 2 [0.1] | 0 | |
| Infections and infestations | ||||||
| Gastrointestinal infection | 3 (8.3) | 3 [0.0] | 1 (6.7) | 1 [0.0] | 0 | |
| Influenza | 4 (11.1) | 4 [0.1] | 1 (6.7) | 1 [0.0] | 0 | |
| Nasopharyngitis | 12 (33.3) | 22 [0.3] | 0 | 3 (30.0) | 6 [0.3] | |
| Pharyngitis | 3 (8.3) | 3 [0.0] | 1 (6.7) | 2 [0.1] | 0 | |
| Rhinitis | 2 (5.6) | 2 [0.0] | 2 (13.3) | 2 [0.1] | 0 | |
| Upper respiratory tract infection | 5 (13.9) | 7 [0.1] | 2 (13.3) | 4 [0.1] | 2 (20.0) | 4 [0.2] |
| Investigations | ||||||
| D-dimer level increased | 9 (25.0) | 12 [0.2] | 1 (6.7) | 1 [0.0] | 1 (10.0) | 1 [0.1] |
| Prothrombin fragments 1 + 2 increased | 7 (19.4) | 12 [0.2] | 2 (13.3) | 2 [0.1] | 0 | |
| Musculoskeletal and connective tissue disorders | ||||||
| Arthralgia | 4 (11.1) | 6 [0.1] | 3 (20.0) | 3 [0.1] | 0 | |
| Back pain | 5 (13.9) | 7 [0.1] | 0 | 0 | ||
| Nervous system disorders | ||||||
| Headache | 8 (22.2) | 12 [0.2] | 1 (6.7) | 1 [0.0] | 1 (10.0) | 1 [0.1] |
| Respiratory, thoracic, and mediastinal disorders | ||||||
| Cough | 4 (11.1) | 4 [0.1] | 0 | 0 | ||
%, percentage of patients with adverse event. AESI, AE of special interest; E, number of AEs; PT, preferred term; R, rate calculated as the number of AEs per patient-years of exposure; SOC, system organ class.
Injection site reactions included injection site bruising, injection site hematoma and injection site hemorrhage.
Bleeding episodes.
The estimated ABRs during the main + extension parts at the last dose level were 4.8 (95% confidence interval [CI], 3.2-7.2) and 6.4 (95% CI, 4.1-9.9) in explorer4 and explorer5, respectively (median ABR, 3.6 and 3.8). Details of the estimated ABR over the entire exposure period are presented in supplemental Table 1. The estimated ABR at last dose level according to hemophilia type in explorer4 was 3.8 (95% CI, 2.2-6.4) for HAwI and 6.2 (95% CI, 3.4-11.1) for HBwI (Figure 3). Estimated spontaneous ABR at the last dose level was 1.8 (95% CI, 1.2-2.6) in explorer 4 and 2.1 (95% CI, 1.3-3.3) in explorer5. In explorer4, spontaneous ABR was 1.6 (95% CI, 1.0-2.7) for patients with HAwI and 2.1 (95% CI, 1.2-3.7) for patients with HBwI. Estimated joint ABR at last dose level was 3.2 (95% CI, 2.1-4.8) and 5.2 (95% CI, 3.1-8.9) in explorer4 and explorer5 (with joint ABRs of 2.7 [95% CI, 1.6-4.6] and 3.8 [95% CI, 2.1-7.0] in patients with HAwI and HBwI in the explorer4 trial).
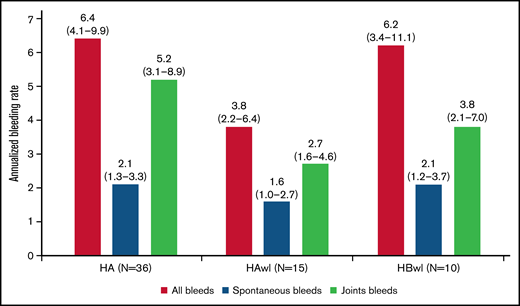
ABRs (treated bleeding episodes on the last concizumab dose level) during the main + extension parts of the phase 2 concizumab trials explorer4 (HAwI, HBwI) and explorer5 (HA) by hemophilia subtype; full analysis set. Least-squares mean estimates of ABR (95% CI) are shown. The ABR estimate was based on a negative binomial regression with log of exposure time as offset and hemophilia type as factor (explorer4).
ABRs (treated bleeding episodes on the last concizumab dose level) during the main + extension parts of the phase 2 concizumab trials explorer4 (HAwI, HBwI) and explorer5 (HA) by hemophilia subtype; full analysis set. Least-squares mean estimates of ABR (95% CI) are shown. The ABR estimate was based on a negative binomial regression with log of exposure time as offset and hemophilia type as factor (explorer4).
In patients who were switched from rFVIIa on-demand during the main part to concizumab prophylaxis during the extension part of explorer4 (n = 8), estimated ABR at the last dose level decreased from 18.6 (95% CI, 12.9-26.9) to a level comparable to that observed in the overall population (4.9; 95% CI, 2.2-10.6) (supplemental Table 2). Similarly, estimated mean spontaneous and joint bleed ABR decreased from 16.9 (95% CI, 11.2-25.5) to 2.5 (95% CI, 1.0-6.2), and 13.8 (95% CI, 9.6-19.9) to 2.9 (95% CI, 1.1-7.7), respectively, in these patients. Three of these patients experienced zero bleeds on their last concizumab dose level (exposure time, 212, 115, and 303 days).21
Pharmacokinetics and pharmacodynamics.
Similar levels of concizumab exposure were observed in patients with and without inhibitors during the main + extension parts of explorer4 and explorer5, and levels of free TFPI were lowered with increasing concizumab concentration (Figure 4A-B). TFPI levels were similarly reduced in patients with HA, HAwI, and HBwI.
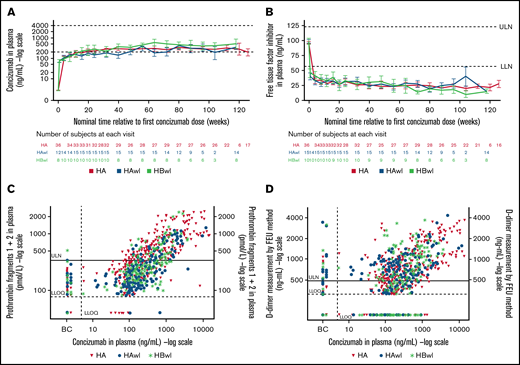
Plots by hemophilia subtype of (A) concizumab plasma concentration (geometric mean) vs time; (B) free TFPI plasma concentration (mean) vs time; (C) prothrombin fragments 1 + 2 vs concizumab plasma concentration; and (D) D-dimers vs concizumab plasma concentration in the main + extension parts of the phase 2 concizumab trials explorer4 (HAwI, HBwI) and explorer5 (HA). All data from visit 16 were allocated to 118 weeks (HAwI and HBwI) or 126 weeks (HA) after first concizumab dose (panels A and B). Data below the lower limit of quantification (LLOQ) were included as LLOQ/2 in the calculations. Horizontal dashed lines in panel A indicate a concizumab exposure level between 200 and 4000 ng/mL. Baseline values for panels C and D were assigned to Visit 9 (last treatment during main part) for the on-demand treatment arm in explorer4. BC, baseline concizumab; FEU, fibrinogen-equivalent units; LLN, lower limit of normal; ULN, upper limit of normal.
Plots by hemophilia subtype of (A) concizumab plasma concentration (geometric mean) vs time; (B) free TFPI plasma concentration (mean) vs time; (C) prothrombin fragments 1 + 2 vs concizumab plasma concentration; and (D) D-dimers vs concizumab plasma concentration in the main + extension parts of the phase 2 concizumab trials explorer4 (HAwI, HBwI) and explorer5 (HA). All data from visit 16 were allocated to 118 weeks (HAwI and HBwI) or 126 weeks (HA) after first concizumab dose (panels A and B). Data below the lower limit of quantification (LLOQ) were included as LLOQ/2 in the calculations. Horizontal dashed lines in panel A indicate a concizumab exposure level between 200 and 4000 ng/mL. Baseline values for panels C and D were assigned to Visit 9 (last treatment during main part) for the on-demand treatment arm in explorer4. BC, baseline concizumab; FEU, fibrinogen-equivalent units; LLN, lower limit of normal; ULN, upper limit of normal.
Mean peak TG potential was within the normal reference range (26-147 nmol/L) for all 3 dose levels before last dose administration at 76 weeks of concizumab treatment in both trials (supplemental Table 3), with similar trends observed for other TG parameters (endogenous thrombin potential, velocity index; data not shown).
Safety
Adverse events.
The majority of AEs reported during the main + extension parts of explorer4 and explorer5 were mild, with no deaths and no events leading to withdrawal from the trial (Table 2). There were no TEs recorded during either trial, including after administration of rFVIIa together with concizumab during the explorer4 trial. Fifteen injection site reactions were reported in 4 patients (40%) with HBwI and 3 reactions in 2 patients (13.3%) with HAwI during the main + extension parts of explorer4, and 25 injection site reactions were reported in 14 patients (39%) during explorer5. The majority of these were bruising, hematoma, or hemorrhage, with most occurring in patients receiving a concizumab dose of 0.15 mg/kg (supplemental Table 4).
No hypersensitivity reactions were observed during the main + extension parts of the explorer4 inhibitor trial. As previously reported by Shapiro et al,17 three nonserious mild events of hypersensitivity reactions were reported in 1 patient (on a dose of concizumab 0.15 mg/kg) during the main part of explorer5. This patient had previously experienced similar symptoms while receiving treatment with another drug and recovered within the same day without requiring treatment for these symptoms. Elevated D-dimer and prothrombin fragment 1 + 2 levels were observed across all concizumab dose levels, indicating the hemostatic effect of concizumab (Figure 4C-D).
A post hoc subanalysis of patients who switched from rFVIIa on-demand during the main part of explorer4 to concizumab prophylaxis during the extension part showed that safety observations in these patients were similar to those seen in the overall study population.
Immunogenicity.
Concizumab ADAs developed in 6 and 9 patients during the main + extension parts of explorer4 and explorer5, respectively, with no apparent clinical effect in all but 1 patient from explorer4. Titers were low in 5 of the patients with ADAs in explorer4 and dropped below detectable levels during the course of the trial in 3 of these patients. Titers were also low in all 9 patients who developed ADAs during explorer5, with 7 patients developing transient ADAs during the trial and 2 developing ADAs at the end-of-trial visit (with no further follow-up in these patients, as per protocol). One patient from explorer4 with initially low-titer ADAs went on to develop a high titer with in vitro neutralizing activity after experiencing severe trauma with a temporary pause in concizumab treatment. This patient continued to receive concizumab, despite free TFPI restoration, and reported 2 bleeding episodes over a period of >7 months. The patient was eventually withdrawn from the trial due to suspicion of no therapeutic effect with restoration of free TFPI; however, the clinical effect of ADAs in this patient remains inconclusive. Further details of the patient’s clinical course are provided in the supplemental Materials. For 1 patient who developed ADAs during explorer5, a positive in vitro neutralizing result was observed at 1 visit, with a negative result at all subsequent visits.22
Discussion
As previously reported, clinical proof-of-concept for once-daily subcutaneous prophylaxis with concizumab across hemophilia subtypes was established during the main parts of the phase 2 explorer4 and explorer5 trials.17 Results from the main + extension parts reported here support the clinical proof-of-concept established in the main parts of the trials and provide details of the longer term (at least 76 weeks) efficacy and safety with concizumab prophylaxis in patients with HA, HAwI, and HBwI.
Concizumab efficacy was maintained, with more patients having a dose escalation during the extension parts of both trials, and estimated ABRs at last concizumab dose level after at least 76 weeks of treatment were comparable to those observed in the main parts (≥24 weeks). As previously reported,17 during the main part of the explorer4 trial, patients with HAwI and HBwI who received concizumab treatment experienced reduced ABRs compared with historical values, whereas those who received rFVIIa had ABRs similar to historical levels. In addition, when these latter patients were switched to concizumab prophylaxis during the extension part, ABR was substantially reduced, with a ratio of 0.26 (95% CI, 0.13-0.54). The explorer4 trial included 10 patients with HBwI, representing a vulnerable, rare patient population with a particularly high level of unmet needs. The results reported here are particularly important for this group, for whom an adequate prophylactic treatment does not currently exist.
Results from the main + extension parts showed that concizumab prophylaxis was generally well tolerated in patients with and without inhibitors, with no AEs leading to withdrawal, TEs, or deaths during either trial. The majority of injection site reactions reported were bruising, hemorrhages, and hematomas, and they mainly occurred in patients on a dose of concizumab 0.15 mg/kg. Concizumab was also well tolerated during the extension part in patients who received rFVIIa on-demand during the main part of explorer4.
Although there were no TEs during explorer4 or explorer5, they are an important safety consideration that must be kept in mind for ongoing development of non-factor replacement therapies. Development of the anti-TFPI monoclonal antibody BAY1093884 was terminated after 3 TEs occurred in 24 patients who received at least 1 dose of treatment.14 This antibody targets the Kunitz 1 (K1) and K2 domains of TFPI,23 meaning there is no free K1 in TFPI bound to the antibody and subsequently no TFPI inhibition of TF-FVIIa, resulting in a potentially higher risk for TEs compared with antibodies that only bind the K2 domain of TFPI. Three cases of thrombotic microangiopathy and 7 TEs were reported during the HAVEN trials with emicizumab treatment, with the thrombotic microangiopathies and 2 of the TEs occurring in patients who had also been exposed to activated prothrombin complex concentrate at doses >100 U/kg per day.24,25 A phase 2 trial evaluating the short interfering RNA conjugate fitusiran (which targets the AT3 gene) was temporarily paused after a drug-related TE and subsequent death. The clinical development program has since been resumed with lower doses of fitusiran and reduced doses of concomitant factor products for the treatment of breakthrough bleeds.13,26 As discussed in the current article, concizumab clinical development was paused after the occurrence of TEs in 3 patients in the phase 3 trials; however, these trials have since been restarted with risk mitigation in place.19
Development of ADAs (inhibitors) is a key limitation of factor replacement therapy for patients with hemophilia. However, ADAs can also be a limitation for non-factor replacement therapies.27,28 In the explorer4 and explorer5 trials, longer term treatment with concizumab exhibited an acceptable immunogenicity profile. Although 25% of patients developed ADAs during the main + extension parts, the majority were low titer and transient with no observed clinical effects, with the exception of 1 patient from explorer4. This patient developed high-titer ADAs with in vitro neutralizing activity after an event of severe trauma. The increase in the titer of ADAs in this patient could potentially be explained by the hypothesis that traumatic injuries can shift the balance of the immune system towards pro-inflammatory and counter-inflammatory phenotypes29 ; however, the clinical impact of ADAs for this patient remains inconclusive due to their complex clinical course.
Concizumab prophylaxis was associated with improvements in patient-reported outcomes during explorer4 and explorer5, particularly in clinically relevant domains such as physical function, vitality, and bodily pain.30 Although improvements were observed in both patient populations, margins of improvement were greater in patients with inhibitors, which may reflect the lower quality of life at baseline in these patients. A key consideration when assessing improvements in quality of life in patients with hemophilia is the impact of treatment on arthropathy.31 The improvements observed in physical functioning domains during the phase 2 concizumab trials may reflect a potential positive impact on arthropathy, and they merit further investigation in future clinical trials.
Overall, the results presented here from the extension parts of the phase 2 explorer4 and explorer5 trials were consistent with main part results and provide support for the development of concizumab as a once-daily, subcutaneous treatment across all hemophilia subtypes. The ongoing explorer7 and explorer8 phase 3 clinical trials will provide further insight into the safety and efficacy of concizumab as a treatment for patients with hemophilia.
Acknowledgments
The authors thank the patients, their families, and all trial investigators for their participation and support in explorer4 and explorer5, and also the external independent data monitoring committee for their assistance with these studies.
These studies were funded by Novo Nordisk A/S. Medical writing support, under the direction of the authors, was provided by Ashfield MedComms GmbH (an Ashfield Health company) and was funded by Novo Nordisk A/S.
Authorship
Contribution: All authors were investigators in either the explorer4 or the explorer5 trial, recruited patients into the studies, analyzed and interpreted the data, contributed to subsequent writing and review of the manuscript, and approved the final version for submission.
Conflict-of-interest disclosure: A.D.S. has received research/grant support from Agios, BioMarin, Bioverativ, Daiichi Sankyo, Genentech/Roche, Glover Blood Therapeutics, Kedrion Biopharma, Novartis, Novo Nordisk, Octapharma, Pfizer, ProMetic BioTherapeutics, Sigilon, and Takeda; has acted as a consultant for Catalyst BioSciences, Genentech/Roche, Kedrion Biopharma, Novo Nordisk, Pfizer, ProMetic BioTherapeutics, and Sigilon; and has participated on the speakers bureau for Genentech/Roche and Novo Nordisk. A.P.W. has acted as a consultant for BioMarin, Novo Nordisk, Shire, UniQure, Spark, Sanofi, Takeda, and HEMA Biologics. G.B. has received speaker fees from Novo Nordisk, Takeda, and BMS. G.C. has acted as a speaker at satellite symposia and during scientific meetings for Novo Nordisk, Bayer, Grifols, LFB, Roche, Sobi, Takeda, Werfen, and Kedrion; has received research funding directly to his institution from CSL Behring, Pfizer, and Sobi; and has participated in advisory board meetings (within the last 2 years) for Novo Nordisk, Ablynx, Alexion, Bayer, Takeda, CSL Behring, Pfizer, Roche, Sanofi, Sobi, and uniQure. G.Y. has received consulting fees from ApcinteX, BioMarin, Genentech/Roche, Grifols, Novo Nordisk, Pfizer, Rani, Sanofi Genzyme, Spark, and Takeda; and has received funds for research support from Genentech/Roche, Grifols, and Takeda. H.E. has received research support or funding from Bayer Vital, CSL Behring, Sobi, and Pfizer; and has acted as a consultant for Bayer Vital, Biotest, CSL Behring, Roche, Sobi, Novo Nordisk, and Pfizer. J.A. has received research grants from Sobi, CSL Behring, Takeda/Shire, and Bayer; and has received speaker fees and acted as a consultant for Octapharma, Novo Nordisk, Pfizer, Bayer, Sobi, Sanofi, CSL Behring, Takeda/Shire, BioMarin, UniQure, and Spark Therapeutics. J.O. has received research support/acted as an investigator for Bayer, Biotest, CSL Behring, Octapharma, Pfizer, Sobi and Takeda; and acted as a consultant, speaker, or advisory board member for/received honoraria from Bayer, Biogen Idec, BioMarin, Biotest, Chugai, CSL Behring, Freeline, Grifols, Novo Nordisk, Octapharma, Pfizer, Roche, Sanofi, Sparks, Sobi, and Takeda. K.K. has participated in advisory board meetings for Novo Nordisk, Takeda, Roche, Bayer, CSL Behring, and Pfizer. L.H.P. has received funding for attending congresses and meetings from Bayer, Novo Nordisk, Pfizer, and Sobi. P.A. has received research funding from Novo Nordisk for clinical trials in hemophilia. P.C. has served on advisory boards for Bayer, Boehringer Ingelheim, CSL Behring, Chugai, Freeline, Novo Nordisk, Pfizer, Roche, Sanofi, Spark, Sobi, and Takeda; and received research funding from Bayer, CSL Behring, Freeline, Novo Nordisk, Pfizer, Sobi, and Takeda. S.Z.-S. has received reimbursement for attending symposia/congresses and honoraria from Baxter, Bayer, Novo Nordisk, Octapharma, Roche, Sobi, and Takeda. T.M. has served on advisory boards for Takeda, Bayer, Novo Nordisk, Chugai, and Pfizer; received educational and investigational support from Chugai and Novo Nordisk; and received honoraria from Takeda, Bayer, Sanofi, Chugai, CSL Behring, JB, KM Biologics, Kirin, Novo Nordisk, Octapharma, and Sysmex. V.J.-Y. has received reimbursement for attending symposia/congresses and/or honoraria for speaking or consulting, and/or research funding from Takeda, Bayer, CSL Behring, Grifols, Novo Nordisk, Sobi, Roche, Octapharma, and Pfizer.
Correspondence: Amy D. Shapiro, Indiana Hemophilia & Thrombosis Center, 8326 Naab Rd, Indianapolis, IN 46260; e-mail: ashapiro@IHTC.org.

