

Key Points
The combination of daratumumab with ATRA is safe but has limited activity in patients with daratumumab-refractory MM.
The limited efficacy may be partially explained by the transient increase in CD38 expression upon ATRA treatment.

Abstract
The efficacy of daratumumab depends partially on CD38 expression on multiple myeloma (MM) cells. We have previously shown that all-trans retinoic acid (ATRA) upregulates CD38 expression and reverts daratumumab-resistance ex vivo. We therefore evaluated the optimal dose, efficacy, and safety of daratumumab combined with ATRA in patients with daratumumab-refractory MM in a phase 1/2 study (NCT02751255). In part A of the study, 63 patients were treated with daratumumab monotherapy. Fifty patients with daratumumab-refractory MM were subsequently enrolled in part B and treated with daratumumab (reintensified schedule) combined with ATRA until disease progression. The recommended phase 2 dose of ATRA in combination with daratumumab was defined as 45 mg/m2. At this dose, the overall response rate (ORR) was 5%, indicating that the primary endpoint (ORR ≥15%) was not met. However, most patients (66%) achieved at least stable disease. After a median follow-up of 43 months, the median progression-free survival (PFS) for all patients was 2.8 months. Patients who previously achieved at least a partial response or minimal response/stable disease with prior daratumumab monotherapy had a significantly longer PFS compared with patients who immediately progressed during daratumumab as single agent (median PFS 3.4 and 2.8 vs 1.3 months). The median overall survival was 19.1 months. The addition of ATRA did not increase the incidence of adverse events. Flow cytometric analysis revealed that ATRA temporarily increased CD38 expression on immune cell subsets. In conclusion, the addition of ATRA and reintensification of daratumumab had limited activity in patients with daratumumab-refractory MM, which may be explained by the transient upregulation of CD38 expression. This trial was registered at www.clinicaltrials.gov as #NCT02751255.
Introduction
The CD38-targeting antibody daratumumab has a favorable toxicity profile and potent single-agent activity in patients with relapsed and/or refractory multiple myeloma (RRMM), with an overall response rate (ORR) of 29% to 37% and median progression-free survival (PFS) of approximately 4 months.1-5 Combinations of daratumumab with immunomodulatory drugs (IMiDs), proteasome inhibitors (PIs), and/or alkylating agents further improve clinical outcomes.6-9 However, approved treatment options for patients with RRMM who become refractory to IMiDs, PIs, and CD38 antibodies are currently limited and the prognosis is poor,10 indicating the unmet medical need for new treatment options in these patients with heavily pretreated MM.
Next to its classic Fc-dependent immune effector functions,11 daratumumab has direct effects by inhibiting CD38 enzymatic activity11 as well as immunomodulatory properties by depleting CD38+ immune suppressor cells, resulting in expansion and enhanced cytotoxic capacity of T cells.12-14 The efficacy of daratumumab depends partially on the expression of CD38 on the tumor cell surface.15,16 Interestingly, all-trans retinoic acid (ATRA) increased CD38 expression on tumor cells and increased ex vivo daratumumab-mediated antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity, even in daratumumab-resistant MM cells.16 In addition, ATRA enhanced the activity of daratumumab in a humanized MM mouse model.16
We therefore evaluated the efficacy and safety of daratumumab combined with ATRA in patients with heavily pretreated daratumumab-refractory MM in the phase 1/2 daratumumab-ATRA study (NCT02751255). We also evaluated the impact of daratumumab and ATRA on the frequency of immune cell subsets and their CD38 expression in sequentially obtained peripheral blood (PB) samples.
Materials and methods
Study design
This study was a prospective, investigator-initiated, nonrandomized, multicenter, open-label, phase 1 dose-finding trial, followed by a phase 2 expansion at the recommended phase 2 dose (RP2D) to evaluate the safety and efficacy of daratumumab and ATRA in RRMM. Both phase 1 and phase 2 consisted of two parts (A and B). Patients were initially treated with daratumumab monotherapy in part A. In case of daratumumab-refractory disease, defined as insufficient response or progression (for definitions, see below), patients were subsequently treated in part B with daratumumab combined with ATRA. The study was conducted in 7 hospitals in The Netherlands. Approval was obtained from the institutional medical ethical committee in each participating center, and the study was performed in accordance with the Declaration of Helsinki. All participants provided written informed consent.
Study objectives
The primary objective of part B of the phase 1 study was to identify the maximum tolerated dose and RP2D of ATRA (out of 3 evaluated dose levels: 15 mg/m2, 30 mg/m2, and 45 mg/m2) in combination with daratumumab. The secondary objective was to evaluate safety and toxicity. The primary objective of part B of the phase 2 study was to assess the ORR (defined as partial response [PR] or better) of daratumumab in combination with ATRA. Secondary objectives of part B were to evaluate the clinical benefit rate (defined as minimal response [MR] or better), the disease control rate (defined as stable disease [SD] or better),17-19 toxicity profile, and PFS and overall survival (OS) in patients treated with daratumumab in combination with ATRA at the RP2D (PFS-B and OS-B). We also assessed the ORR and safety profile of daratumumab monotherapy (part A) and determined PFS from start of daratumumab monotherapy to progression during daratumumab combined with ATRA (PFS-AB) and OS from start of daratumumab monotherapy (OS-AB).
Study population
Patients were eligible for enrollment in part A of the trial (daratumumab monotherapy) if they had MM, relapsed from or refractory to at least 2 prior lines of treatment, and measurable disease, defined as any of the following: (1) serum monoclonal protein ≥5 g/L; (2) urine monoclonal protein ≥200 mg/24 h; or (3) serum immunoglobulin free light chain ≥100 mg/L and abnormal serum κ to λ free light chain ratio. Previous therapy with daratumumab or other anti-CD38 therapies was allowed but not within 6 months before initiation of study treatment. Additional inclusion and exclusion criteria are listed in the supplemental Methods.
Patients were subsequently enrolled in part B of the study (daratumumab combined with ATRA) in case of insufficient response to daratumumab monotherapy (defined as disease progression during the first cycle of treatment in part A, less than MR after the second treatment cycle, or less than PR after the third treatment cycle), or in case of disease progression after an initial response (PR or better). Inclusion criteria for part B were similar to part A, except for a creatinine clearance of ≥30 mL/min.
Drug administration
Daratumumab was administered intravenously at a dose of 16 mg/kg according to the approved schedule, which consists of 8 weekly infusions, then 8 biweekly infusions, and monthly infusions thereafter.1,2 In part B, ATRA was orally administered twice daily during 3 days per each daratumumab infusion, whereby daratumumab was administered on the third day (supplemental Table 1). Upon enrollment in part B of the study, daratumumab dosing was resumed according to the aforementioned administration schedule, starting again with 8 weekly infusions. Daratumumab in combination with ATRA was continued until disease progression.
Safety and efficacy assessments
Adverse events (AEs) were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03.20 All AEs grade ≥2 were assessed from inclusion until 30 days after the last administration of any study drug. In addition, all-grade infusion-related reactions and polyneuropathy were reported. Treatment response was assessed at the end of each cycle according to the International Myeloma Working Group Uniform Response Criteria.17
Statistics
The phase 2 study was designed to determine whether treatment with daratumumab in combination with ATRA at the RP2D warrants further investigation in clinical trials. We expected that in part B of the phase 2 study, retreatment with daratumumab alone in this daratumumab-unresponsive population would result in an ORR close to 0% (0% to 2%). We determined that for daratumumab combined with ATRA in this patient category, an ORR <15% would be unacceptable. To have power 1 – β = 0.90 to detect an improvement, with 2-sided significance level α = 0.05, a total of 40 patients treated at the RP2D of ATRA would be required. All analyses were performed according to the intention-to-treat principle. PFS was defined as time from registration until progression or death, whichever came first. OS was defined as time from registration until death from any cause. Patients still alive at date of last follow-up were censored. PFS and OS were estimated using the Kaplan-Meier method. Differences between survival curves in subgroup analyses were tested for statistical significance using the 2-sided log-rank test. Unless otherwise specified, the analyses for part B included all 8 patients treated at the RP2D (dose level 3) in phase 1 and all 36 patients treated in phase 2. Comparisons between continuous variables were performed using Wilcoxon matched-pairs signed rank test. P values <0.05 were considered significant. All statistical analyses were performed using SPSS software (version 22) or GraphPad prism (version 8). Clinical data were monitored by the Clinical Research Bureau (Amsterdam University Medical Center).
Details and additional methods are presented in the supplemental Data.
Results
Patient characteristics
A total of 63 patients were enrolled in this phase 1/2 study from July 2016 to October 2017 and initially treated with daratumumab monotherapy (part A; Figure 1). Median number of prior lines of treatment was 4 (range 2-11; Table 1). All patients were exposed to lenalidomide and a PI; 89% were IMiD refractory, 71% were PI refractory, and 67% were double refractory to both an IMiD and a PI. Three patients had previously been treated with daratumumab monotherapy in the GEN501 study1 16, 42, and 48 months before study enrollment, with as best response MR in 1 patient and very good PR (VGPR) in 2 patients (Table 1). At least PR was achieved in 41% of the patients treated in part A, with VGPR in 14% and complete remission in 5%. Best response in the 3 patients who had previously been exposed to daratumumab was PR in 1 patient and progressive disease (PD) in 2 patients.
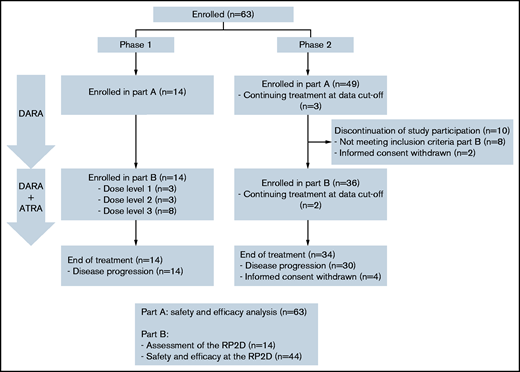
Patient flow diagram. Of 63 patients enrolled in part A (daratumumab monotherapy), 2 patients were still on treatment at data cutoff, 14 patients were enrolled in part B (daratumumab + ATRA) phase 1, 36 patients were enrolled in part B phase 2, and 10 patients were not enrolled in part B because of ineligibility or withdrawal of informed consent. All 63 patients enrolled in part A were evaluated for safety and efficacy, all 14 patients enrolled in part B phase 1 were evaluated for the assessment of the RP2D, and all 44 patients treated at the RP2D in part B were evaluated for safety and efficacy.
Patient flow diagram. Of 63 patients enrolled in part A (daratumumab monotherapy), 2 patients were still on treatment at data cutoff, 14 patients were enrolled in part B (daratumumab + ATRA) phase 1, 36 patients were enrolled in part B phase 2, and 10 patients were not enrolled in part B because of ineligibility or withdrawal of informed consent. All 63 patients enrolled in part A were evaluated for safety and efficacy, all 14 patients enrolled in part B phase 1 were evaluated for the assessment of the RP2D, and all 44 patients treated at the RP2D in part B were evaluated for safety and efficacy.
Baseline characteristics
| Characteristic | Part A (n = 63) | Part B phase 1 (n = 14) | Part B phase 2 (n = 36) | |||
|---|---|---|---|---|---|---|
| Median age, y (range) | 69 (38-80) | 70 (47-79) | 70 (46-80) | |||
| <65, n (%) | 20 (31.7) | 5 (35.7) | 10 (27.8) | |||
| 65 to <75, n (%) | 30 (47.6) | 6 (42.9) | 15 (41.7) | |||
| ≥75, n (%) | 13 (20.6) | 3 (21.4) | 11 (30.6) | |||
| Sex, n (%) | ||||||
| Female | 24 (38.1) | 4 (28.6) | 17 (47.2) | |||
| Male | 39 (61.9) | 10 (71.4) | 19 (52.8) | |||
| WHO performance score, n (%) | ||||||
| 0 | 29 (46.0) | 8 (57.1) | 15 (41.7) | |||
| 1 | 25 (39.7) | 5 (35.7) | 16 (44.4) | |||
| 2 | 7 (11.1) | 1 (7.1) | 5 (13.9) | |||
| Unknown | 2 (3.2) | 0 | 0 | |||
| Extramedullary plasmacytomas, n (%) | ||||||
| No | 53 (84.1) | 12 (85.7) | 32 (88.9) | |||
| Yes | 8 (12.7) | 2 (14.3) | 3 (8.3) | |||
| Type of monoclonal heavy chain, n (%) | ||||||
| IgG | 36 (57.1) | 4 (28.6) | 26 (72.2) | |||
| IgA | 7 (11.1) | 1 (7.1) | 5 (13.9) | |||
| Light chain only | 13 (20.6) | 6 (42.9) | 3 (8.3) | |||
| Bence-Jones | 7 (11.1) | 3 (21.4) | 2 (5.6) | |||
| Type of light chain, n (%) | ||||||
| Kappa | 40 (63.5) | 2 (14.3) | 29 (80.6) | |||
| Lambda | 23 (36.5) | 12 (85.7) | 7 (19.4) | |||
| Time since start first treatment, median years (range) | 6.8 (1.5-16.0) | 7.1 (2.5-16.0) | 5.5 (1.9-14.8) | |||
| Prior lines of treatment, median (range) | 4 (2-11) | 8 (4-11) | 5 (3-12) | |||
| >3 prior lines, n (%) | 41 (65.1) | 14 (100) | 32 (88.9) | |||
| Autologous SCT, n (%) | 38 (60.3) | 10 (71.4) | 18 (50.0) | |||
| Allogeneic SCT, n (%) | 8 (12.7) | 2 (14.3) | 3 (8.3) | |||
| Double-class refractory, n (%)* | 42 (66.7) | 10 (71.4) | 27 (75.0) | |||
| Triple-class refractory, n (%)† | 2 (3.2) | 10 (71.4) | 27 (75.0) | |||
| Prior IMID, n (%) | Exposed | Refractory‡ | Exposed | Refractory‡ | Exposed | Refractory‡ |
| Thalidomide | 32 (50.8) | 11 (17.5) | 9 (64.3) | 5 (35.7) | 16 (44.4) | 5 (13.9) |
| Lenalidomide | 63 (100) | 51 (81.0) | 14 (100) | 11 (78.6) | 36 (100) | 27 (75.0) |
| Pomalidomide | 23 (36.5) | 22 (34.9) | 6 (42.9) | 6 (42.9) | 13 (36.1) | 13 (36.1) |
| Prior PI, n (%) | Exposed | Refractory‡ | Exposed | Refractory‡ | Exposed | Refractory‡ |
| Bortezomib | 61 (96.8) | 37 (58.7) | 14 (100) | 10 (71.4) | 34 (94.4) | 24 (66.7) |
| Carfilzomib | 7 (11.1) | 7 (11.1) | 0 | 0 | 5 (13.9) | 5 (13.9) |
| Ixazomib | 2 (3.2) | 2 (3.2) | 0 | 0 | 2 (5.6) | 2 (5.6) |
| Prior monoclonal antibody, n (%) | Exposed | Refractory‡ | Exposed | Refractory‡ | Exposed | Refractory‡ |
| Daratumumab | 3 (4.8)§ | 2 (3.2) | 14 (100) | 14 (100) | 36 (100) | 36 (100) |
| Elotuzumab | 2 (3.2) | 2 (3.2) | 0 | 0 | 2 (5.6) | 2 (5.6) |
| Durvalumab | 6 (9.5) | 6 (9.5) | 0 | 0 | 6 (16.7) | 6 (16.7) |
| ISS at registration, n (%) | ||||||
| I | 14 (22.2) | 5 (35.7) | 8 (22.2) | |||
| II | 30 (47.6) | 2 (14.3) | 22 (61.1) | |||
| III | 14 (22.2) | 6 (42.9) | 2 (5.6) | |||
| Unknown | 5 (7.9) | 1 (7.1) | 4 (11.1) | |||
| Cytogenetic abnormalities¶, n (%) | ||||||
| t(4;14) | 1 (1.6) | 0 | 0 | |||
| t(14;16) | 1 (1.6) | 1 (7.1) | 0 | |||
| t(14;20) | 0 | 0 | 0 | |||
| del(17p) | 11 (17.5) | 2 (14.3) | 3 (8.3) | |||
| amp(1q) | 25 (39.7) | 7 (50.0) | 13 (36.1) | |||
| del(1p) | 5 (7.9) | 3 (21.4) | 2 (5.6) | |||
| del(13q) | 17 (27) | 7 (50.0) | 7 (19.4) | |||
| Cytogenetic risk profile#, n (%) | ||||||
| High-risk | 36 (57.1) | 8 (57.1) | 18 (50.0) | |||
| Standard-risk | 14 (22.2) | 2 (14.3) | 11 (30.6) | |||
| Not available | 13 (20.6) | 4 (28.6) | 7 (19.4) | |||
| Laboratory values at baseline, median (range) | ||||||
| Absolute neutrophil count, ×109/L | 2.92 (0.60-8.87) | 2.89 (1.67-8.00) | 3.36 (0.90-8.27) | |||
| Hemoglobin level, mmol/L | 7.0 (4.8-8.9) | 7.1 (5.0-8.4) | 7.3 (6.0-9.3) | |||
| Platelet count, ×109/L | 168 (34-479) | 146 (50-445) | 181 (32-600) | |||
| Creatinine, µmol/L | 88 (53-228) | 91 (61-140) | 88 (62-140) | |||
| Calcium, mmol/L** | 2.41 (2.05-2.95) | 2.35 (2.19-2.56) | 2.42 (2.15-2.60) | |||
| LDH, U/L | 204 (119-1356) | 199 (145-279) | 179 (142-641) | |||
| Bone marrow plasma cell percentage in biopsy (median, range)†† | 50 (0-100) | NA | NA | |||
| Characteristic | Part A (n = 63) | Part B phase 1 (n = 14) | Part B phase 2 (n = 36) | |||
|---|---|---|---|---|---|---|
| Median age, y (range) | 69 (38-80) | 70 (47-79) | 70 (46-80) | |||
| <65, n (%) | 20 (31.7) | 5 (35.7) | 10 (27.8) | |||
| 65 to <75, n (%) | 30 (47.6) | 6 (42.9) | 15 (41.7) | |||
| ≥75, n (%) | 13 (20.6) | 3 (21.4) | 11 (30.6) | |||
| Sex, n (%) | ||||||
| Female | 24 (38.1) | 4 (28.6) | 17 (47.2) | |||
| Male | 39 (61.9) | 10 (71.4) | 19 (52.8) | |||
| WHO performance score, n (%) | ||||||
| 0 | 29 (46.0) | 8 (57.1) | 15 (41.7) | |||
| 1 | 25 (39.7) | 5 (35.7) | 16 (44.4) | |||
| 2 | 7 (11.1) | 1 (7.1) | 5 (13.9) | |||
| Unknown | 2 (3.2) | 0 | 0 | |||
| Extramedullary plasmacytomas, n (%) | ||||||
| No | 53 (84.1) | 12 (85.7) | 32 (88.9) | |||
| Yes | 8 (12.7) | 2 (14.3) | 3 (8.3) | |||
| Type of monoclonal heavy chain, n (%) | ||||||
| IgG | 36 (57.1) | 4 (28.6) | 26 (72.2) | |||
| IgA | 7 (11.1) | 1 (7.1) | 5 (13.9) | |||
| Light chain only | 13 (20.6) | 6 (42.9) | 3 (8.3) | |||
| Bence-Jones | 7 (11.1) | 3 (21.4) | 2 (5.6) | |||
| Type of light chain, n (%) | ||||||
| Kappa | 40 (63.5) | 2 (14.3) | 29 (80.6) | |||
| Lambda | 23 (36.5) | 12 (85.7) | 7 (19.4) | |||
| Time since start first treatment, median years (range) | 6.8 (1.5-16.0) | 7.1 (2.5-16.0) | 5.5 (1.9-14.8) | |||
| Prior lines of treatment, median (range) | 4 (2-11) | 8 (4-11) | 5 (3-12) | |||
| >3 prior lines, n (%) | 41 (65.1) | 14 (100) | 32 (88.9) | |||
| Autologous SCT, n (%) | 38 (60.3) | 10 (71.4) | 18 (50.0) | |||
| Allogeneic SCT, n (%) | 8 (12.7) | 2 (14.3) | 3 (8.3) | |||
| Double-class refractory, n (%)* | 42 (66.7) | 10 (71.4) | 27 (75.0) | |||
| Triple-class refractory, n (%)† | 2 (3.2) | 10 (71.4) | 27 (75.0) | |||
| Prior IMID, n (%) | Exposed | Refractory‡ | Exposed | Refractory‡ | Exposed | Refractory‡ |
| Thalidomide | 32 (50.8) | 11 (17.5) | 9 (64.3) | 5 (35.7) | 16 (44.4) | 5 (13.9) |
| Lenalidomide | 63 (100) | 51 (81.0) | 14 (100) | 11 (78.6) | 36 (100) | 27 (75.0) |
| Pomalidomide | 23 (36.5) | 22 (34.9) | 6 (42.9) | 6 (42.9) | 13 (36.1) | 13 (36.1) |
| Prior PI, n (%) | Exposed | Refractory‡ | Exposed | Refractory‡ | Exposed | Refractory‡ |
| Bortezomib | 61 (96.8) | 37 (58.7) | 14 (100) | 10 (71.4) | 34 (94.4) | 24 (66.7) |
| Carfilzomib | 7 (11.1) | 7 (11.1) | 0 | 0 | 5 (13.9) | 5 (13.9) |
| Ixazomib | 2 (3.2) | 2 (3.2) | 0 | 0 | 2 (5.6) | 2 (5.6) |
| Prior monoclonal antibody, n (%) | Exposed | Refractory‡ | Exposed | Refractory‡ | Exposed | Refractory‡ |
| Daratumumab | 3 (4.8)§ | 2 (3.2) | 14 (100) | 14 (100) | 36 (100) | 36 (100) |
| Elotuzumab | 2 (3.2) | 2 (3.2) | 0 | 0 | 2 (5.6) | 2 (5.6) |
| Durvalumab | 6 (9.5) | 6 (9.5) | 0 | 0 | 6 (16.7) | 6 (16.7) |
| ISS at registration, n (%) | ||||||
| I | 14 (22.2) | 5 (35.7) | 8 (22.2) | |||
| II | 30 (47.6) | 2 (14.3) | 22 (61.1) | |||
| III | 14 (22.2) | 6 (42.9) | 2 (5.6) | |||
| Unknown | 5 (7.9) | 1 (7.1) | 4 (11.1) | |||
| Cytogenetic abnormalities¶, n (%) | ||||||
| t(4;14) | 1 (1.6) | 0 | 0 | |||
| t(14;16) | 1 (1.6) | 1 (7.1) | 0 | |||
| t(14;20) | 0 | 0 | 0 | |||
| del(17p) | 11 (17.5) | 2 (14.3) | 3 (8.3) | |||
| amp(1q) | 25 (39.7) | 7 (50.0) | 13 (36.1) | |||
| del(1p) | 5 (7.9) | 3 (21.4) | 2 (5.6) | |||
| del(13q) | 17 (27) | 7 (50.0) | 7 (19.4) | |||
| Cytogenetic risk profile#, n (%) | ||||||
| High-risk | 36 (57.1) | 8 (57.1) | 18 (50.0) | |||
| Standard-risk | 14 (22.2) | 2 (14.3) | 11 (30.6) | |||
| Not available | 13 (20.6) | 4 (28.6) | 7 (19.4) | |||
| Laboratory values at baseline, median (range) | ||||||
| Absolute neutrophil count, ×109/L | 2.92 (0.60-8.87) | 2.89 (1.67-8.00) | 3.36 (0.90-8.27) | |||
| Hemoglobin level, mmol/L | 7.0 (4.8-8.9) | 7.1 (5.0-8.4) | 7.3 (6.0-9.3) | |||
| Platelet count, ×109/L | 168 (34-479) | 146 (50-445) | 181 (32-600) | |||
| Creatinine, µmol/L | 88 (53-228) | 91 (61-140) | 88 (62-140) | |||
| Calcium, mmol/L** | 2.41 (2.05-2.95) | 2.35 (2.19-2.56) | 2.42 (2.15-2.60) | |||
| LDH, U/L | 204 (119-1356) | 199 (145-279) | 179 (142-641) | |||
| Bone marrow plasma cell percentage in biopsy (median, range)†† | 50 (0-100) | NA | NA | |||
Double-class refractory is defined as both IMiD and PI refractory disease.
Triple-class refractory is defined as IMID, PI, and CD38-targeting antibody refractory disease.
Refractory disease is defined as PD during therapy, no response (<PR), or PD within 60 days of stopping treatment, according to the International Uniform Response Criteria for Multiple Myeloma.
3 patients had previously been treated with daratumumab 16, 42, and 48 months before registration.
As determined by fluorescence in situ hybridization or single nucleotide polymorphism array on purified MM cells before start of daratumumab treatment.
According to the criteria proposed by Sonneveld et al. Blood 2016.46 High risk disease is defined by the presence of t(4;14), t(14;16), t(14;20), del(17/17p), and/or gain(1q).
Calcium corrected for serum albumin using the following formula: corrected calcium = calcium measured + ((40 − albumin) × 0.02).
Based on data from 39 patients (62%).
At data cutoff (1 October 2020), 50 of these 63 patients were subsequently treated with daratumumab and ATRA in part B. Fourteen patients were enrolled in the phase 1 dose-finding part, and 36 patients were enrolled in part B after the RP2D for ATRA was determined. Reasons for enrolling in part B were insufficient response to daratumumab monotherapy, defined as PD after the first cycle (n = 6 [12%]), less than MR after the second cycle (n = 16 [32%]), less than PR after the third cycle (n = 8 [16%]), and disease progression after initial response to daratumumab monotherapy (n = 20 [40%]). Thirteen of the 63 patients were not enrolled in part B: 3 patients were still treated in part A of the study (not yet eligible for part B), 8 did not meet eligibility criteria for part B, and 2 withdrew informed consent.
Dose escalation
Fourteen patients (Table 1; Figure 1) were enrolled in part B of the phase 1 study. These 14 patients were treated with daratumumab combined with ATRA at one of the 3 dose levels (15, 30, and 45 mg/m2). In the absence of dose limiting toxicity, the maximum tolerated dose was not reached (supplemental Table 2), and the RP2D for ATRA in combination with daratumumab was defined as 45 mg/m2.
Efficacy of daratumumab combined with ATRA at the RP2D
Thirty-six patients (Table 1; Figure 1) were enrolled in part B of the phase 2 study at the time of insufficient response or progression during daratumumab monotherapy. For the efficacy analysis, we also included the 8 patients treated at the RP2D in part B of phase 1 of the study. Most of these 44 patients (73%) had triple-class refractory MM at the time of enrollment in part B. At data cutoff, 2 patients were still treated with daratumumab combined with ATRA and 42 patients discontinued treatment, with disease progression as the most common reason (38 patients [86%]).
At least PR was observed in 2 of these 44 patients (5%). Both patients achieved a VGPR in part B, they had achieved MR and complete remission as best response in part A and were enrolled in part B 2 and 16 months after start of daratumumab monotherapy. The clinical benefit rate (MR or better) was 9%, and the disease control rate (SD or better) was 66% (Figure 2A). After a median follow-up of 43 months, the median PFS of daratumumab combined with ATRA (PFS-B) was 2.8 months (95% CI, 1.6-4.0 months) and the median OS (OS-B) was 19.1 months (95% CI, 15.0-23.1 months) (Figure 2B-C). Overall, median PFS from the start of daratumumab monotherapy followed by the ATRA combination (PFS-AB) was 7.9 months (95% CI, 4.9-10.8 months) and median OS (OS-AB) 28.2 months (95% CI, 11.9-44.6 months) (Figure 2D-E).
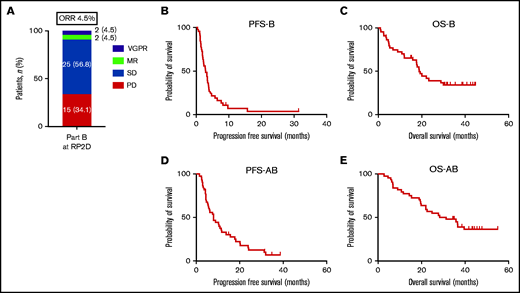
Response, PFS, and OS for patients treated with daratumumab combined with ATRA at the RP2D. (A) Response, (B) PFS-B, and (C) OS-B for all patients treated with daratumumab combined with ATRA at the RP2D of 45 mg/m2. (D) PFS-AB and (E) OS-AB for the whole treatment strategy of daratumumab monotherapy followed by the daratumumab/ATRA combination for all patients treated at the RP2D.
Response, PFS, and OS for patients treated with daratumumab combined with ATRA at the RP2D. (A) Response, (B) PFS-B, and (C) OS-B for all patients treated with daratumumab combined with ATRA at the RP2D of 45 mg/m2. (D) PFS-AB and (E) OS-AB for the whole treatment strategy of daratumumab monotherapy followed by the daratumumab/ATRA combination for all patients treated at the RP2D.
The best response achieved during daratumumab monotherapy in part A had an impact on PFS in part B. PFS-B was superior in patients who had either achieved PR or better (median PFS-B 3.4 months; 95% CI, 3.0-3.8; P = .002) or SD/MR (median PFS-B 2.8 months; 95% CI, 1.1-4.6; P = .027), compared with patients who immediately progressed during daratumumab monotherapy in part A (median PFS-B 1.3 months; 95% CI, 1.0-1.7; Figure 3A). Median OS-B was 19.1 months (95% CI, 15.5-22.7) in patients with PR or better, 19.4 months (95% CI, 11.8-26.9) in patients with SD/MR, and 17.9 months (95% CI, 0-48.8) in patients with PD in part A. Median PFS-AB was significantly longer for patients who had achieved either PR or better (median PFS-AB 18.2 months; 95% CI, 12.4-23.9; P = .001) or SD/MR (median PFS-AB 5.4 months; 95% CI, 3.8-7.0; P = .002), compared with patients who immediately progressed during daratumumab monotherapy in part A (median PFS-AB 2.5 months; 95% CI, 2.1-2.9; Figure 3B). Median OS-AB was 36.4 months (95% CI, 29.2-43.6) in patients with PR or better, 21.8 months (95% CI, 15.8-27.8) in patients with SD/MR, and 19.5 months (95% CI, 0-48.5) in patients with PD in part A.
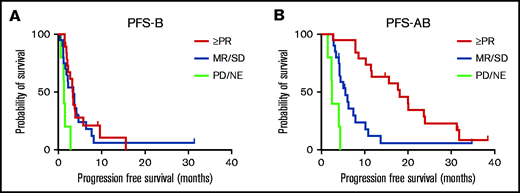
PFS-B and PFS-AB stratified according to best response to daratumumab monotherapy. (A) PFS-B and (B) PFS-AB stratified according to best response to daratumumab monotherapy during part A of the study (PR or better, SD/MR, or PD). NE, not evaluable.
PFS-B and PFS-AB stratified according to best response to daratumumab monotherapy. (A) PFS-B and (B) PFS-AB stratified according to best response to daratumumab monotherapy during part A of the study (PR or better, SD/MR, or PD). NE, not evaluable.
Safety of daratumumab combined with ATRA at the RP2D
All 63 patients enrolled in part A of the study were evaluated for hematologic and nonhematologic AEs occurring during daratumumab monotherapy. The AE profile observed in part A was similar to what has been reported in other RRMM studies with daratumumab as single agent (Table 2).1,2 AEs did not result in treatment discontinuation.
Incidence and severity of adverse events
| Part A n = 63 | Part B RDL n = 44 | |||
|---|---|---|---|---|
| Events | Grade 2 n (%) | Grade 3-4 n (%) | Grade 2 n (%) | Grade 3-4 n (%) |
| Hematologic | ||||
| Anemia | 11 (17) | 8 (13)* | 21 (48) | 3 (7)* |
| Thrombocytopenia | 9 (14) | 11 (17)† | 3 (7) | 6 (14)† |
| Neutropenia | 13 (21) | 9 (14)‡ | 4 (9) | 5 (11)‡ |
| Febrile neutropenia | — | 1 (2)‡ | — | — |
| Infusion-related reaction | 12 (19)§ | — | — | — |
| Infections | 26 (42) | 11 (18) | 28 (64) | 6 (14) |
| Upper respiratory tract | 14 (22) | — | 14 (32) | — |
| Pneumonia | 3 (5) | 4 (6) | 2 (5) | 5 (11) |
| Gastrointestinal | 2 (3) | 3 (5) | 1 (2) | — |
| Urinary tract infection | 1 (2) | — | 7 (16) | 1 (2) |
| Skin infection | 4 (6) | 1 (2) | 1 (2) | — |
| Other | 2 (3)¶ | 3 (5)# | 3 (7)** | — |
| Headache | 1 (2) | — | 5 (11) | — |
| Thromboembolic event | — | 1 (2) | — | — |
| Second primary malignancy | 2 (3)†† | — | — | — |
| Fatigue | 6 (10) | 1 (2) | 3 (7) | 1 (2) |
| Part A n = 63 | Part B RDL n = 44 | |||
|---|---|---|---|---|
| Events | Grade 2 n (%) | Grade 3-4 n (%) | Grade 2 n (%) | Grade 3-4 n (%) |
| Hematologic | ||||
| Anemia | 11 (17) | 8 (13)* | 21 (48) | 3 (7)* |
| Thrombocytopenia | 9 (14) | 11 (17)† | 3 (7) | 6 (14)† |
| Neutropenia | 13 (21) | 9 (14)‡ | 4 (9) | 5 (11)‡ |
| Febrile neutropenia | — | 1 (2)‡ | — | — |
| Infusion-related reaction | 12 (19)§ | — | — | — |
| Infections | 26 (42) | 11 (18) | 28 (64) | 6 (14) |
| Upper respiratory tract | 14 (22) | — | 14 (32) | — |
| Pneumonia | 3 (5) | 4 (6) | 2 (5) | 5 (11) |
| Gastrointestinal | 2 (3) | 3 (5) | 1 (2) | — |
| Urinary tract infection | 1 (2) | — | 7 (16) | 1 (2) |
| Skin infection | 4 (6) | 1 (2) | 1 (2) | — |
| Other | 2 (3)¶ | 3 (5)# | 3 (7)** | — |
| Headache | 1 (2) | — | 5 (11) | — |
| Thromboembolic event | — | 1 (2) | — | — |
| Second primary malignancy | 2 (3)†† | — | — | — |
| Fatigue | 6 (10) | 1 (2) | 3 (7) | 1 (2) |
Patients requiring erythrocyte transfusions: part A, n = 16 (25%); part B, n = 11 (25%).
Patients requiring platelet transfusion: part A, n = 6 (10%); part B, n = 4 (9%).
Patients requiring granulocyte colony-stimulating factor support: part A, n = 12 (19%); part B, n = 6 (14%).
Any grade infusion-related reaction: 22 patients (35%).
Eye infection and tooth infection (both n = 1).
Cytomegalovirus infection, peripherally inserted central catheter line infection and tooth infection (all n = 1).
Candida stomatitis, sinusitis, and no focus found (all n = 1).
Basal cell carcinoma and mesothelioma (both n = 1).
All 44 patients treated with daratumumab combined with ATRA at the RP2D were evaluated for AEs from start of treatment until 30 days after the last daratumumab infusion (Table 2). The frequency of hematological toxicity and infectious complications was comparable to what was observed with daratumumab monotherapy in part A (Table 2). ATRA-related headache was reported in 5 patients (11%; all grade 2), mainly during the first 2 cycles, and was manageable with acetaminophen. One patient developed grade 2 noncardiac chest pain after initiation of ATRA treatment, which resolved after ATRA administration was interrupted on day 4 of the first cycle. The chest pain did not reoccur after rechallenge with ATRA on day 8 of the first cycle. No infusion-related reactions occurred after the addition of ATRA and reintensification of daratumumab. All toxicities could be successfully managed with supportive care, and no treatment discontinuations because of AEs were reported.
Subsequent therapy
At data cutoff, 42 of 44 patients treated with daratumumab and ATRA at the RP2D had discontinued treatment. Thirty-eight of these 42 patients (90%) received subsequent anti-MM therapy. An IMiD-containing regimen without a PI was the most common first subsequent therapy (26 patients [68%]; mostly pomalidomide-based (24 patients [63%]). This was followed by treatment with the cereblon E3 ligase modulator (CELMoD) iberdomide (10 patients [26%]; Table 3). The ORR and median PFS on the subsequent line of therapy were 61% and 6.2 months (95% CI, 2.2-10.3 months), respectively. In later lines of therapy, 5 patients received bispecific antibodies and 2 chimeric antigen receptor (CAR) T-cell therapies.
Subsequent therapy
| All patients receiving subsequent treatment (n = 38) | |
|---|---|
| IMiD-based regimen, n (%) | |
| Pomalidomide-dexamethasone | 15 (39) |
| Pomalidomide-cyclophosphamide-dexamethasone | 6 (16) |
| Pomalidomide-prednisone | 2 (5) |
| Lenalidomide-cyclophosphamide-prednisone | 2 (5) |
| CELMoD-based regimen, n (%) | |
| Iberdomide ± dexamethasone | 10 (26) |
| PI-based regimen, n (%) | |
| Bortezomib-cyclophosphamide-dexamethasone | 1 (3) |
| Carfilzomib-dexamethasone | 1 (3) |
| IMiD + monoclonal antibody, n (%) | |
| Elotuzumab-pomalidomide-dexamethasone | 1 (3) |
| All patients receiving subsequent treatment (n = 38) | |
|---|---|
| IMiD-based regimen, n (%) | |
| Pomalidomide-dexamethasone | 15 (39) |
| Pomalidomide-cyclophosphamide-dexamethasone | 6 (16) |
| Pomalidomide-prednisone | 2 (5) |
| Lenalidomide-cyclophosphamide-prednisone | 2 (5) |
| CELMoD-based regimen, n (%) | |
| Iberdomide ± dexamethasone | 10 (26) |
| PI-based regimen, n (%) | |
| Bortezomib-cyclophosphamide-dexamethasone | 1 (3) |
| Carfilzomib-dexamethasone | 1 (3) |
| IMiD + monoclonal antibody, n (%) | |
| Elotuzumab-pomalidomide-dexamethasone | 1 (3) |
Impact of daratumumab and ATRA on frequency of immune cell subsets and their CD38 expression levels
We assessed the effect of daratumumab and ATRA on frequency and CD38 expression of immune cell subsets in sequential PB samples. In accordance with prior studies, daratumumab monotherapy (part A) significantly decreased the absolute number of PB natural killer (NK) cells (P < .0001; supplemental Figure 1A).12,21 Directly after addition of ATRA, we observed a modest further reduction in NK cell numbers (P = .0176; supplemental Figure 1B). Similar to total NK cells, activated NK cells (CD16+CD56dim) were reduced by daratumumab and remained low during the whole study period. In accordance with our previous findings,12 there was an increase in T cells after initiation of daratumumab monotherapy, which persisted throughout the study as well as after the addition of ATRA (supplemental Figure 2). Daratumumab with or without ATRA did not affect B-cell numbers. Addition of ATRA to daratumumab resulted in a modest but significant decrease in monocytes in the first cycle (P = .0002), with recovery to baseline at day 3 of cycle 2 (supplemental Figure 1A-B).
Daratumumab monotherapy rapidly reduced CD38 expression levels on monocytes, B cells, T cells, and NK cells (Figure 4A). Three days after initiation of ATRA, CD38 expression significantly increased on these immune cell subsets (Figure 4B). Although ATRA treatment was continued, cell surface expression levels of CD38 on B, T, and NK cells on day 3 of cycle 2 and at the time of progression were again similar to what we observed before initiation of combined ATRA and daratumumab treatment (Figure 4B). For monocytes, CD38 cell surface expression on day 3 of cycle 2 was lower compared with day 3 cycle 1 but still higher than before initiation of ATRA treatment. However, at the time of progression, CD38 cell surface expression on monocytes was again similar to what was observed before initiation of ATRA treatment (Figure 4B).
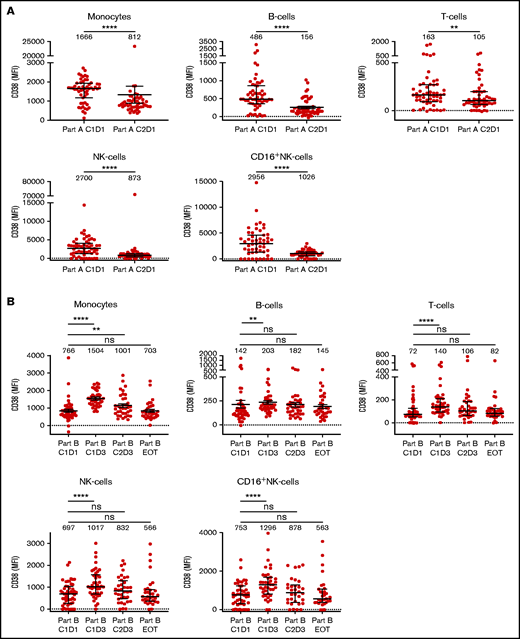
CD38 expression levels on immune cell subsets in PB samples obtained during treatment with daratumumab monotherapy followed by daratumumab with ATRA. (A) CD38 expression levels on monocytes, B cells, T cells, NK cells, and activated NK cells (CD16+) in sequential PB samples obtained before start of daratumumab monotherapy (C1D1 of part A; n = 55), and after 1 cycle of daratumumab monotherapy (C2D1 of part A; n = 51). (B) CD38 expression levels on these immune cell subsets in sequential PB samples obtained before start of ATRA treatment (C1D1 of part B; n = 51), before first dose of daratumumab after initiation of ATRA (C1D3 of part B; n = 42), after 1 cycle of daratumumab and ATRA treatment (C2D3 of part B; n = 36), and at disease progression during daratumumab and ATRA treatment (EOT, part B; n = 33). Dots represent individual expression levels; error bars represent median and IQR. Median fluorescence intensity is provided for each timepoint. Differences between indicated groups were calculated using Wilcoxon matched-pairs rank test. *P < .05; **P < .01; ***P < .001; ****P < .0001. EOT, end of treatment; IQR, interquartile range; MFI, median fluorescence intensity; ns, not significant.
CD38 expression levels on immune cell subsets in PB samples obtained during treatment with daratumumab monotherapy followed by daratumumab with ATRA. (A) CD38 expression levels on monocytes, B cells, T cells, NK cells, and activated NK cells (CD16+) in sequential PB samples obtained before start of daratumumab monotherapy (C1D1 of part A; n = 55), and after 1 cycle of daratumumab monotherapy (C2D1 of part A; n = 51). (B) CD38 expression levels on these immune cell subsets in sequential PB samples obtained before start of ATRA treatment (C1D1 of part B; n = 51), before first dose of daratumumab after initiation of ATRA (C1D3 of part B; n = 42), after 1 cycle of daratumumab and ATRA treatment (C2D3 of part B; n = 36), and at disease progression during daratumumab and ATRA treatment (EOT, part B; n = 33). Dots represent individual expression levels; error bars represent median and IQR. Median fluorescence intensity is provided for each timepoint. Differences between indicated groups were calculated using Wilcoxon matched-pairs rank test. *P < .05; **P < .01; ***P < .001; ****P < .0001. EOT, end of treatment; IQR, interquartile range; MFI, median fluorescence intensity; ns, not significant.
Discussion
Here, we show that in patients with triple-class exposed daratumumab-refractory MM (73% triple-class refractory), the combination of daratumumab and ATRA at the RP2D is safe, with a toxicity profile similar to daratumumab monotherapy. Although 66% of patients achieved SD or better, the primary endpoint was not met with an ORR of 5%.
The limited efficacy of adding ATRA to daratumumab may be explained by our findings that ATRA increased CD38 expression on immune cells but did not restore CD38 to baseline levels. In addition, the increase was only temporary, which may be related to ongoing transfer of CD38/daratumumab complexes from target cells to monocytes or granulocytes (trogocytosis).21 Selection of cells with lower CD38 levels may also contribute to the temporary increase in CD38 expression. In our study, patients received ATRA during a 3-day course per daratumumab administration to prevent toxicity related to CD38 upregulation in normal tissues. Therefore, we cannot exclude that continuous ATRA treatment might be a more effective strategy. In addition, patients who have not yet been treated with a CD38 antibody may experience more benefit from ATRA than patients with daratumumab-refractory MM. It would also be interesting to evaluate whether the efficacy of daratumumab-based triplet regimens (including an IMiD or PI)6-8 can be further improved by the addition of ATRA. Next to ATRA, histone deacytelase inhibitors and ruxolitinib, which target the JAK/STAT3 pathway, have also been shown to increase CD38 expression on MM cells, resulting in enhanced ex vivo activity of daratumumab.22-24 To date, no data are available from clinical trials exploring these combinations.
CD38 downregulation is not the only mechanism of resistance to CD38 antibodies.25 There is evidence that NK-cell depletion,26 increased levels of complement inhibitors,15 upregulation of CD47,27 adhesive interactions with stromal cells,28 and T-cell exhaustion29 may also contribute to daratumumab resistance. Because of these pleiotropic modes of resistance, a strategy that is focused solely on CD38 upregulation may have only modest effects. In this respect, several other clinical studies are evaluating different strategies to reverse resistance against CD38 antibodies, including the adoptive transfer of CD38 knockout NK cells30,31 and blockade of the PD-1/PD-L1 axis at the time of development of CD38 antibody–refractory disease.32-34 Another strategy, explored in an ongoing study, is retreatment with daratumumab after a treatment-free interval of at least 3 months,35 which allows for (partial) recovery of CD38 expression levels and NK cells.15,26 A recent study showed that single-agent isatuximab, a different CD38-targeting antibody, was not effective after the development of daratumumab resistance.36 This may be explained by the partly overlapping mode of action.37,38 As a result of the similarities between both antibodies, it is unlikely that the combination of isatuximab and ATRA will be able to reverse daratumumab resistance.
Although clinical activity of daratumumab-ATRA was limited, there may be a role for this combination in a select patient population in the absence of alternative treatment options. First, the combination of daratumumab and ATRA might be considered for heavily pretreated patients who progress during daratumumab monotherapy and had previously achieved PR or better. In our study, these patients achieved a median PFS of 3.4 months, which is comparable to what can be achieved with other options available outside clinical trials, such as retreatment with drugs used in prior lines (PR or better in 31% with median PFS of 3.4 months in the MAMMOTH study),10 or use of the recently approved drugs selinexor, belantamab mafodotin, and melflufen (PR or better in 26% to 31% and median PFS of 2.9-3.9 months in patients with triple-class refractory MM).39-41 Addition of ATRA to daratumumab can also be considered in case of suboptimal response (MR or SD) during daratumumab monotherapy. These patients had a median PFS of 5.4 months for the whole treatment strategy. Although there was no head-to-head comparison with daratumumab monotherapy until progression, these data compare favorably to the median PFS of 3.0 months for patients who achieved SD/MR and continued daratumumab monotherapy until progression (pooled analysis of the GEN501 and Sirius studies).5
At the time of enrollment in part B of the study, all patients were triple-class exposed and daratumumab-refractory (73% triple-class refractory). Notably, median OS of these patients was 19.1 months. This compares favorably to what is reported for patients with CD38 antibody–refractory disease in the MAMMOTH study (79% triple-class refractory; median OS of 8.6 months)10 and may be related to increasing access to novel active immunotherapeutic modalities in clinical trials, such as CELMoDs, bispecific antibodies, and CAR T cells. In addition, daratumumab treatment may have a beneficial effect on subsequent therapies,42 which can be partially explained by the continued presence of daratumumab in the circulation during the first months after the last infusion, resulting from the approximately 21-day half-life of daratumumab, which allows for synergistic interactions with IMiD or CELMoD agents.14,43,44 The immunomodulatory effects of daratumumab may also contribute to the promising response rate with subsequent immune-stimulatory therapies.12
A limitation of our study is that as a consequence of the trial design, we cannot differentiate between the impact of ATRA and daratumumab intensification on clinical outcomes. However, although anecdotal, data do not suggest that response can be regained by daratumumab intensification alone.45 In addition, we did not assess the effect of ATRA on CD38 expression on MM cells in bone marrow samples but studied CD38 expression on PB immune cell subsets as a less invasive alternative. Importantly, we have previously shown that daratumumab-mediated changes in CD38 expression on immune cells reflect changes observed on MM cells.21
In conclusion, the addition of ATRA and reintensification of daratumumab were safe but had limited activity in patients with daratumumab-refractory MM, which may be partially explained by the transient increase in CD38 expression.
Acknowledgments
The authors thank Jhon A. Marin Soto, Paola Homan-Weert, and Inoka Twickler, who helped measure the peripheral blood samples, the research nurses and study coordinators at each of the participating sites, the members of the independent data monitoring committee for evaluating the data, and the patients who participated in this trial and their families.
This work was supported by grants from Janssen Research & Development.
Authorship
Contribution: N.W.C.J.v.d.D. and K.A.F. designed the study and interpreted results; M.C.M., M.-D.L., A.B., G.M.J.B., M.J.K., S.K.K., I.S.N., S.Z., P.S., and N.W.C.J.v.d.D. enrolled patients and provided patient material; K.A.F., P.W.C.M.-B., C.P.M.V., and T.M. analyzed data and interpreted results; and K.A.F. and N.W.C.J.v.d.D. wrote the first draft of the manuscript; all authors helped critically review the manuscript and checked the final version of it.
Conflict-of-interest disclosure: M.C.M. holds a consultancy or advisory role for Gilead Sciences, BMS, Alnylam, Janssen Cilag, Takeda, and Servier (all paid to employer) and hospitality from Celgene; A.B. received honoraria from Celgene, Janssen, Amgen, and Takeda; M.J.K. received research support from Kite/Gilead and honoraria from Kite/Gilead, Novartis, BMS/Celgene, Takeda, Roche, and Miltenyi Biotec (all paid to institution); T.M. received research support from Janssen Pharmaceuticals; S.Z. received honoraria from Celgene, Sanofi, Takeda, and Janssen and research funding from Celgene, Takeda, and Janssen; P.S. received honoraria from Amgen, BMS, Celgene, Janssen, Karyopharm, and Takeda and receives research funding from Amgen, Celgene, Janssen, Karyopharm, SkylineDx, and Takeda; N.W.C.J.v.d.D. received research support from Janssen Pharmaceuticals, AMGEN, Celgene, Novartis, Cellectis, and BMS and serves on advisory boards for Janssen Pharmaceuticals, AMGEN, Celgene, BMS, Takeda, Roche, Novartis, Bayer, Adaptive, and Servier. The remaining authors declare no competing financial interestst.
Correspondence: Niels W. C. J. van de Donk, Department of Hematology, Amsterdam UMC, Vrije Universiteit Amsterdam, De Boelelaan 1117, 1081 HV Amsterdam, The Netherlands; e-mail: n.vandedonk@amsterdamumc.nl.

