

Key Points
CDAR achieved 95% CR and 80% negative MRD, durable up to 10 years.
Carrying TP53 mutation and not achieving negative MRD after CDAR were associated with shorter survival outcomes.

Abstract
Hairy cell leukemia variant (HCLv) responds poorly to purine analogue monotherapy. Rituximab concurrent with cladribine (CDAR) improves response rates, but long-term outcomes are unknown. We report final results of a phase 2 study of CDAR for patients with HCLv. Twenty patients with 0 to 1 prior courses of cladribine and/or rituximab, including 8 who were previously untreated, received cladribine 0.15 mg/kg on days 1 to 5 with 8 weekly rituximab doses of 375 mg/m2 beginning day 1. Patients received a second rituximab course ≥6 months after cladribine, if and when minimal residual disease (MRD) was detected in blood. The complete remission (CR) rate from CDAR was 95% (95% confidence interval, 75-100). Sixteen (80%) of 20 patients (95% confidence interval, 56-94) became MRD negative according to bone marrow at 6 months. The median duration of MRD-negative CR was 70.1 months, and 7 of 16 are still MRD negative up to 120 months. With a median follow-up of 69.7 months, 11 patients received delayed rituximab, and the 5-year progression-free survival (PFS) and overall survival (OS) were 63.3% and 73.9%, respectively. Five patients with TP53 mutations had shorter PFS (median, 36.4 months vs unreached; P = .0024) and OS (median, 52.4 months vs unreached; P = .032). MRD-negative CR at 6 months was significantly associated with longer PFS (unreached vs 17.4 months; P < .0001) and OS (unreached vs 38.2 months; P < .0001). Lack of MRD in blood at 6 months was also predictive of longer PFS and OS (P < .0001). After progression following CDAR, median OS was 29.7 months. CDAR is effective in HCLv, with better outcomes in patients who achieve MRD-negative CR. This trial is registered at www.clinicaltrials.gov as #NCT00923013.
Introduction
Hairy cell leukemia variant (HCLv) is a distinct subtype of B-cell leukemia that shares morphologic features with classic HCL but is biologically distinct; it lacks expression of CD25, CD123, tartrate-resistant acid phosphatase, and annexin 1a and is wild type for BRAF, lacking the V600E mutation.1,2 Although early reports indicated that HCLv is only 10% as common as HCL,3,4 a 2016 study estimated 810 new HCLv cases vs 1100 new HCL cases in the United States.5
HCLv responds very poorly to single-agent purine analogue and has worse overall survival (OS) compared with HCL.6 There is no prospective study evaluating purine analogue monotherapy for HCLv; however, the complete remission (CR) rate from first-line cladribine was only 7% based on retrospective studies.3,7-11 The response rate and duration become lower and shorter with repeated purine analogue therapy, and median OS from diagnosis is <6 to 9 years depending on risk factors, compared with >25 years for classic HCL.3,12,13 The TP53 abnormality is relatively common,14 suggesting a relationship to chemoresistant disease, but this theory has never been analyzed in a prospective trial.
Rituximab combined with purine analogue can improve response rate and duration. In a phase 2 study at the MD Anderson Cancer Center, 7 patients newly diagnosed with HCLv received 8 weekly doses of rituximab 1 month after completion of cladribine, and the CR rate was 86%.15 With a median follow-up of 60.2 months, 5-year failure-free survival and OS were 64.3% and 51.4%, respectively, the latter influenced by several patients with secondary malignancies.15,16 We reported preliminary data from a phase 2 study of cladribine in combination with concurrent rituximab (CDAR) in patients with HCLv.6 In the first 10 patients treated on the trial, the CR rate was 90%, and 8 of 9 patients who achieved CR remained free of minimal residual disease (MRD) according to the most sensitive test, bone marrow aspirate flow cytometry, at a median follow-up duration of 27 months (range, 12-48 months). This analysis reported the high efficacy of CDAR in HCLv but lacked long-term outcomes. Since the prior report, we completed enrollment of 20 patients to the study and have much longer follow-up. We therefore update the results of this phase 2 HCLv trial of CDAR and for the first time show the impact of MRD and TP53 mutation on long-term survival outcomes.
Patients and methods
Eligibility criteria
The study was registered at ClinicalTrials.gov (#NCT00923013); it was performed in accordance with the Declaration of Helsinki and was approved by the National Cancer Institute Institutional Review Board. Patients required a diagnosis of HCLv with 0 to 1 prior courses of cladribine and 0 to 1 prior courses of rituximab. Those who received previous concurrent purine analogue and rituximab were excluded, and patients required treatment due to cytopenias (neutrophils <1 × 109/L, hemoglobin <10 g/dL, or platelet <100 × 109/L), lymphocytosis >5 × 109/L, symptomatic splenomegaly, or recurrent infections. Patients were not excluded for severe cytopenias, and those with infections under treatment were eligible. The clinical protocol also included patients with classic HCL randomized to receive CDAR vs cladribine followed by delayed rituximab when blood became MRD positive. Results from a randomized cohort treating patients with purine analog–naive HCL were recently published,17 and those with one prior purine analogue continue to be randomized in a separate stratification.
Study design and objectives
Patients received cladribine IV 0.15 mg/kg on days 1 to 5 with 8 weekly doses of rituximab IV 375 mg/m2 begun on day 1 (CDAR). Patients received a second course of rituximab, also 8 weekly doses, at least 6 months after the first course if and when blood MRD was positive. According to protocol, delayed rituximab could also be used if HCLv-related cytopenias developed at levels lower than those required for CR, although this did not occur with negative blood flow cytometry in the 20 patients with HCLv.
The primary end point was to determine the CR rate according to CDAR. With 20 evaluable patients, if there are ≥3 CRs in the 20 patients (15%), there is only 7.5% probability that this would occur if the true CR rate was 5%; there is a 90.9% probability that this would occur if the true CR rate was 25%. Secondary end points included evaluating MRD by blood and bone marrow aspirate flow cytometry18 and bone marrow biopsy immunohistochemistry (IHC)19 to determine MRD-negative survival after the first and second courses of rituximab. The detection limit of flow cytometry was 0.002% using markers, including CD19, CD11c, CD20, and CD103. Although bone marrow biopsy IHC and blood flow cytometry were part of the MRD analysis, results of these 2 tests were never positive when bone marrow aspirate flow cytometry was negative, and thus those with negative bone marrow aspirate flow cytometry were the patients with MRD-negative disease.
Bone marrow studies were performed at 4 weeks, 6 months, and yearly for 2.5 years after cladribine treatment and biannually thereafter. Blood flow cytometry was performed at 4 weeks, quarterly until 1 year, and semiannually until 2.5 years after cladribine treatment and yearly thereafter. This schedule would reset for the beginning of delayed rituximab once that was begun, except that bone marrow biopsy was not performed 4 weeks after beginning delayed rituximab. Blood counts were performed approximately twice as frequently as flow cytometry. Patients are being followed up indefinitely on this schedule or until starting next treatment. Response was assessed as previously described,20 except that for partial response (PR), additional established criteria included ≥50% improvements in neutrophils, hemoglobin (≥9 g/dL was sufficient if transfusion dependent at baseline), and platelets, and a ≥50% reduction in circulating HCL cells and lymph nodes.21 PR did not require ≥50% reduction in marrow HCL infiltration so that PR could be assessed without bone marrow, and patients were considered in CR regardless of residual cytopenias if negative for MRD by all tests.
Whole-exome sequencing
Peripheral blood mononuclear cell samples obtained by Ficoll centrifugation containing HCLv cells comprising >95% of total B cells were magnetically labeled with CD19 MicroBeads (Miltenyi Biotec, Auburn, CA) according to the company protocol; they were then positively sorted with an autoMACSTM Separator (Miltenyi Biotec). The genomic DNA was prepared from the sorted cell pellet by using the Quick-DNA/RNA MicroPrep Plus Kit (Zymo Research, Irvine, CA). The genomic DNA sample was randomly fragmented by sonication (Covaris, Woburn, MA) to the size of 180 to 280 bp fragments. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities, and enzymes were removed. After adenylation of 3' ends of DNA fragments, adapter oligonucleotides were ligated. DNA fragments with ligated adapter molecules on both ends were selectively enriched in a polymerase chain reaction (PCR) reaction. After the PCR reaction, the libraries were hybridized in buffer with biotin-labeled probes, and magnetic beads with streptomycin were then used to capture the exons. After washing beads and digesting probes, the captured libraries were enriched in a PCR reaction to add index tags. Products were purified with the AMPure XP system (Beckman Coulter, Beverly, MA) and quantified by using the Agilent high-sensitivity DNA assay on the Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA). Trimmomatic (v.0.38)22 was used to trim the adapters and low-quality bases before aligning reads to the human hg38 reference genome with BWA (v.0.7.17).23 Mapped reads were then de-duplicated with Picard Tools (v.1.119), followed by re-alignment, and base quality score recalibration was performed by using the Genome Analysis Toolkit (GATK v.3.8.0).24
Somatic variant analysis
Somatic variant calling was performed by using muTect (v.1.1.7),25 MuTect2,26 and VarDict (v1.4)27 in tumor-only mode, and only mutations called by at least 2 of the 3 programs were kept. Variants were annotated with Ensembl’s Variant Effect Predictor (VEP v.92)28 and converted to Mutation Annotation Format using the vcf2maf tool (v1.6.16).29 Common variants annotated with a frequency larger than 0.001 in the ExAC, gnomAD, or 1000 Genomes databases, as well as frequently mutated genes30 found in exome cohorts that are likely to be false positive, were removed. In addition, variants with an alternate allele frequency <5%, variant allele depth <5, or total sequencing depth <20× were excluded, and only variants annotated by SnpEff (V4.3)31 as high or moderate for deleteriousness were kept.
Progression-free survival (PFS) was calculated from the beginning of CDAR until loss of CR after delayed rituximab or initiation of next therapy or death from any cause. OS was calculated from the beginning of CDAR until death from any cause. PFS and OS were calculated by using the Kaplan-Meier method, with curves compared using a log-rank test. MRD-negative duration was defined from the date of negative bone marrow flow cytometry until the last assessment of negative or first assessment of relapsed MRD in bone marrow or blood, and the duration of MRD-negative CR was also determined by using the Kaplan-Meier method. The duration could not be extended without bone marrow core negative for IHC and aspirate negative by flow cytometry. Median follow-up was considered the median time from treatment to death or data cutoff for the 20 patients. Median potential follow-up was considered the median time from treatment to data cutoff for the 20 patients. Data cutoff was July 4, 2021. Hazard ratios and 95% confidence intervals (CIs) were calculated by using Cox models. All analyses were performed by using SAS version 9.4 (SAS Institute, Inc, Cary, NC). All P values are two-tailed.
Results
Patient characteristics
Twenty patients were enrolled (Table 1). Median age was 67 years (range, 42-86 years). Seven (37%) of 19 evaluated patients had unmutated IGHV4-34 gene. The median time from diagnosis to CDAR was 12.4 months (range, 1.9-147.7 months). Eight patients were previously untreated; 1 had only splenectomy; 6 had prior cladribine; 1 had prior cladribine and splenectomy; 1 had prior rituximab; 1 had prior rituximab and splenectomy; 1 had prior cladribine, splenectomy, and 2 unsuccessful attempts at completing a dose of rituximab; and 1 had prior combination rituximab-containing chemotherapy (rituximab, cyclophosphamide, vincristine, and prednisone) followed by cladribine.
Patient characteristics at baseline
| Characteristic | Value |
|---|---|
| N | 20 |
| Age, y | 67 (42-86) |
| Male sex, n (%) | 15 (75) |
| White blood cell count, ×109/L | 13.63 (1.14-210.6) |
| Absolute lymphocyte count, ×109/L | 8.33 (0.37-201.6) |
| Absolute neutrophil count, ×109/L | 2.91 (0.63-8.00) |
| Hemoglobin, g/L | 11.7 (7.6-14.8) |
| Platelet count, ×109/L | 119 (49-474) |
| Spleen size, cm | 22.5 (15.5-36) |
| Circulating HCLv cells, ×106/L | 5,877 (4.14-241 000) |
| Bone marrow HCLv cells, % | 35 (5-90) |
| IGHV4-34 unmutated IgH, n (%) | 7 (37) |
| Time from diagnosis to treatment, mo | 12.4 (1.9-147.7) |
| Prior treatment | |
| Untreated | 8 |
| Cladribine monotherapy | 6 |
| Cladribine, rituximab, splenectomy | 1 |
| Cladribine, splenectomy | 1 |
| R-CVP, cladribine | 1 |
| Rituximab, splenectomy | 1 |
| Rituximab monotherapy | 1 |
| Splenectomy | 1 |
| Time from prior treatment, mo | 14.5 (3.5-53.7) |
| Characteristic | Value |
|---|---|
| N | 20 |
| Age, y | 67 (42-86) |
| Male sex, n (%) | 15 (75) |
| White blood cell count, ×109/L | 13.63 (1.14-210.6) |
| Absolute lymphocyte count, ×109/L | 8.33 (0.37-201.6) |
| Absolute neutrophil count, ×109/L | 2.91 (0.63-8.00) |
| Hemoglobin, g/L | 11.7 (7.6-14.8) |
| Platelet count, ×109/L | 119 (49-474) |
| Spleen size, cm | 22.5 (15.5-36) |
| Circulating HCLv cells, ×106/L | 5,877 (4.14-241 000) |
| Bone marrow HCLv cells, % | 35 (5-90) |
| IGHV4-34 unmutated IgH, n (%) | 7 (37) |
| Time from diagnosis to treatment, mo | 12.4 (1.9-147.7) |
| Prior treatment | |
| Untreated | 8 |
| Cladribine monotherapy | 6 |
| Cladribine, rituximab, splenectomy | 1 |
| Cladribine, splenectomy | 1 |
| R-CVP, cladribine | 1 |
| Rituximab, splenectomy | 1 |
| Rituximab monotherapy | 1 |
| Splenectomy | 1 |
| Time from prior treatment, mo | 14.5 (3.5-53.7) |
Data are presented as median (range) unless otherwise indicated. IgH, immunoglobulin heavy chain rearrangement; R-CVP, rituximab, cyclophosphamide, vincristine, and prednisone.
Response status at 4 weeks
To determine response status at 4 weeks after cladribine, patients underwent bone marrow and imaging studies. Fourteen patients (70%) achieved CR at 4 weeks, all with negative bone marrow IHC, and 3 additional patients had negative bone marrow findings by hematoxylin and eosin stain and IHC, but blood counts were not yet consistent with CR. Bone marrow aspirate flow cytometry was negative in 9 (45%), dry-tap in 1 (5%, considered MRD positive), and nonevaluable in 2 (10%) patients. Thus, at 4 weeks, 9 (50%) of 18 evaluable patients achieved MRD-negative CR (95% CI, 26-74), 17 (85%) of 20 had IHC-negative bone marrow biopsy findings, and 13 (68%) of 19 evaluable patients achieved negative MRD in blood.
Response to CDAR at 6 months
By 6 months, 95% (95% CI, 75-100) of patients had achieved a best response of CR. The one patient (#9) who did not achieve CR had by far the largest spleen size (14.7 × 30 × 36 cm) and had prior rituximab therapy. Another patient (#11) with a large spleen (12.2 × 25.7 × 26.4 cm) achieved CR by day 27, negative according to IHC but MRD positive by flow cytometry of blood and bone marrow. This patient’s HCLv rapidly progressed and by the 6-month time point had already relapsed from CR. At 6 months after CDAR, 18 (90%) of 20 patients were in CR; all 18 were MRD negative according to IHC, 16 (80%) were MRD negative according to all tests, and 17 (85%) were MRD negative in blood.
MRD-negative durations before delayed rituximab
Of the 16 patients achieving MRD-negative CR, 9 (56%) became MRD positive with a median MRD-negative CR duration of 70.1 months, and the other 7 patients remain in MRD-negative CR for 21 to 120 months (median, 29.1 months) (Figure 1A). The MRD-negative CR durations in Figure 1A could not be extended without a bone marrow negative for both IHC and flow cytometry, the latter being the most sensitive. Because bone marrow procedures in clinical practice are not frequently performed after treatment, and as blood MRD in this trial was the trigger for delayed rituximab, we also observed blood MRD-negative duration. As shown in Figure 1B, of the 17 patients who became blood MRD-negative after CDAR, median blood MRD-negative survival was 70.0 months. Seven patients currently remain blood MRD negative for 27 to 139 months (median, 59.1 months). These responses and durations were all achieved before and without delayed rituximab.

Survival outcomes and duration of response. MRD-negative CR duration is shown for 16 patients achieving MRD-negative CR with CDAR (A), and blood MRD-negative duration is shown for 17 patients becoming blood MRD-negative (B). OS and PFS are shown for all 20 patients (C), and OS after progression is shown for 8 patients who progressed (D). PFS (E) and OS (F) are shown for 5 patients with a TP53 mutation vs 14 patients without a TP53 mutation.
Survival outcomes and duration of response. MRD-negative CR duration is shown for 16 patients achieving MRD-negative CR with CDAR (A), and blood MRD-negative duration is shown for 17 patients becoming blood MRD-negative (B). OS and PFS are shown for all 20 patients (C), and OS after progression is shown for 8 patients who progressed (D). PFS (E) and OS (F) are shown for 5 patients with a TP53 mutation vs 14 patients without a TP53 mutation.
Delayed rituximab
As shown in Figure 2, a total of 11 patients received delayed rituximab, in all cases triggered by MRD detectable in blood, with a time from CDAR to delayed rituximab of 6 to 82 months (median, 34.8 months). These 11 patients included 9 patients becoming blood MRD positive (Figure 1B) in addition to 2 patients who remained blood MRD positive after CDAR. Figure 2 shows that delayed rituximab led to blood becoming MRD negative in 7 of 11 patients; 5 of these 7 patients have been restaged at 6 months from delayed rituximab and achieved MRD-negative CR with negative bone marrow MRD. Of note, one (#13) of those achieving MRD-negative CR by delayed rituximab had not become MRD negative earlier with CDAR. Three of five who achieved MRD-negative CR after delayed rituximab became MRD positive again after 12, 12, and 36 months, respectively, and the other 2 patients remain MRD negative at 37 and 42 months. Of the 11 patients who received delayed rituximab, 5 patients then received the next line of treatment due to progression, and 1 progressed and died without next treatment; these progressions were cytopenias associated with disease or lymphadenopathy. The median time from delayed rituximab to progression in 6 patients was 13 months (range, 6-30 months).
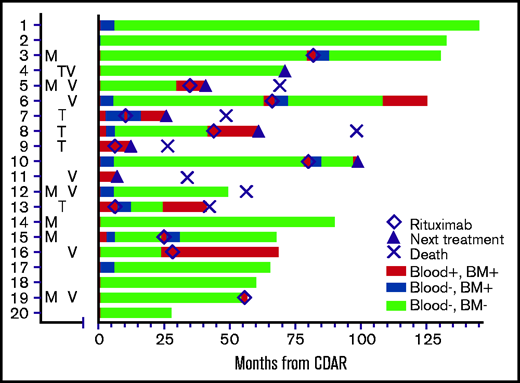
Swimmer’s plot showing MRD status until last assessment and timing of delayed rituximab (◇), next treatment (▴), or death (×). Letters to the left of the swimmer’s plot indicate which patients had mutations in MAP2K (M), TP53 (T), or expressed unmutated IGHV4-34 immunoglobulin rearrangements (V). TP53 and MAP2K mutations are listed in Table 2. Patient #2 was not characterized molecularly. Patient #19 became MRD-negative in blood (blue) after delayed rituximab, just before date cutoff. BM, bone marrow.
Swimmer’s plot showing MRD status until last assessment and timing of delayed rituximab (◇), next treatment (▴), or death (×). Letters to the left of the swimmer’s plot indicate which patients had mutations in MAP2K (M), TP53 (T), or expressed unmutated IGHV4-34 immunoglobulin rearrangements (V). TP53 and MAP2K mutations are listed in Table 2. Patient #2 was not characterized molecularly. Patient #19 became MRD-negative in blood (blue) after delayed rituximab, just before date cutoff. BM, bone marrow.
In addition to 11 patients who received delayed rituximab, 2 patients received alternative chemoimmunotherapy without delayed rituximab due to rapid progression of disease, including massive splenomegaly (patient #4 and patient #11). Thus, 7 patients in total received their next line of treatment beyond delayed rituximab on protocol, 4 with other chemoimmunotherapy, 1 with ibrutinib, 1 with additional rituximab, and 1 with pentostatin. Seven patients have died at last follow-up: 5 with HCLv, 1 from lung cancer but with active HCLv limiting treatment, and 1 with Parkinson’s disease while still in MRD-negative CR.
Response durations
The 5-year PFS was 63.3% (95% CI, 37.9-80.6) as shown in Figure 1C and could be lengthened by response to delayed rituximab. The 5-year OS was 73.9% (95% CI, 48.2-88.2). Ten-year PFS and OS were 44.3% (95% CI, 17.7-68.3) and 57.6% (95% CI, 29.1-78.1), respectively. As shown in Figure 1D, the median time to death from progression was 29.7 months (95% CI, 0-40.6). An association between prior splenectomy or purine analogue and either PFS or OS was not observed (data not shown).
IGHV4-34, TP53, and MAP2K mutation in HCLv
Unmutated IGHV4-34 was seen in 7 patients (#4, #5, #6, #11, #12, #16, and #19). Having IGHV4-34 unmutated HCLv was not associated with either PFS (P = .51) or OS (P = .83). Although this was a post hoc analysis, we performed whole-exome sequencing using available samples and found that 5 (26%) of 19 evaluable patients had TP53 mutations (#4, #7, #8, #9, and #13) (Table 2). Clonal and subclonal status is shown in the variant allele frequency (VAF) column of Table 2. The data are consistent with clonal homozygous TP53 mutations in patients #7, #8, and #13 (VAF ∼1). Patient #9 appears to have 2 clonal heterozygous TP53 mutations on different alleles (VAF ∼0.5). Thus, the wild-type TP53 allele appears absent in these 4 patients who died, whereas patient #4, who is still living, has a subclonal TP53 mutation. Patients with a TP53 mutation had significantly higher risk of MRD-positive disease at 6 months compared with those without a TP53 mutation (3 of 5 vs 1 of 14; P = .037) and had shorter PFS (median, 36.4 months vs unreached; P = .0024) (Figure 1E) and OS (median, 52.4 months vs unreached; P = .032) (Figure 1F).
TP53 and MAP2K mutations
| Patient no. | Gene | Mutation | Sequencing depth | VAF | % of B cells | |
|---|---|---|---|---|---|---|
| Variant | Total | |||||
| 4 | TP53 | c.560-1G>A (p.X187_splice) | 54 | 205 | 0.26 | >99 |
| 7 | TP53 | c.154C>T (p.Q52*) | 59 | 59 | 1.00 | >99 |
| 8 | TP53 | c.517G>A (p.V173M) | 47 | 49 | 0.96 | >99 |
| 9 | TP53 | c.659A>G (p.Y220C) | 63 | 130 | 0.48 | >99 |
| 9 | TP53 | c.470T>C (p.V157A) | 40 | 90 | 0.44 | >99 |
| 13 | TP53 | c.672 + 1G>T (p.X224_splice) | 49 | 52 | 0.94 | >99 |
| 3 | MAP2K1 | c.361T>A (p.C121S) | 126 | 323 | 0.39 | 95 |
| 5 | MAP2K1 | c.362G>C (p.C121S) | 72 | 157 | 0.46 | >99 |
| 12 | MAP2K2 | c.170T>G (p.F57C) | 196 | 444 | 0.44 | >99 |
| 14 | MAP2K1 | c.173A>C(p.Q58P) | 20 | 335 | 0.06 | 93 |
| 14 | MAP2K1 | c.308T>A (p.I103N) | 19 | 191 | 0.10 | 93 |
| 15 | MAP2K1 | c.529C>A (p.L177M) | 34 | 65 | 0.52 | >99 |
| 19 | MAP2K1 | c.169A>G (p.K57E) | 46 | 112 | 0.41 | 96 |
| Patient no. | Gene | Mutation | Sequencing depth | VAF | % of B cells | |
|---|---|---|---|---|---|---|
| Variant | Total | |||||
| 4 | TP53 | c.560-1G>A (p.X187_splice) | 54 | 205 | 0.26 | >99 |
| 7 | TP53 | c.154C>T (p.Q52*) | 59 | 59 | 1.00 | >99 |
| 8 | TP53 | c.517G>A (p.V173M) | 47 | 49 | 0.96 | >99 |
| 9 | TP53 | c.659A>G (p.Y220C) | 63 | 130 | 0.48 | >99 |
| 9 | TP53 | c.470T>C (p.V157A) | 40 | 90 | 0.44 | >99 |
| 13 | TP53 | c.672 + 1G>T (p.X224_splice) | 49 | 52 | 0.94 | >99 |
| 3 | MAP2K1 | c.361T>A (p.C121S) | 126 | 323 | 0.39 | 95 |
| 5 | MAP2K1 | c.362G>C (p.C121S) | 72 | 157 | 0.46 | >99 |
| 12 | MAP2K2 | c.170T>G (p.F57C) | 196 | 444 | 0.44 | >99 |
| 14 | MAP2K1 | c.173A>C(p.Q58P) | 20 | 335 | 0.06 | 93 |
| 14 | MAP2K1 | c.308T>A (p.I103N) | 19 | 191 | 0.10 | 93 |
| 15 | MAP2K1 | c.529C>A (p.L177M) | 34 | 65 | 0.52 | >99 |
| 19 | MAP2K1 | c.169A>G (p.K57E) | 46 | 112 | 0.41 | 96 |
The last column is the percentage of HCLv cells relative to total B cells in the sample.
Although not all patients were tested by using fluorescence in situ hybridization to rule out 17p deletions, plots of polymorphisms as well as missense mutations across chromosome 17 for all 19 patients showed no evidence of an entire arm (17p) missing (data not shown). As shown in Table 2, MAP2K mutations appear to be clonal heterozygous in patients #3, #5, #12, #15, and #19 (VAF ∼0.5) and subclonal in patient #14. MAP2K mutation was not associated with either PFS (P = .41) or OS (P = .72).
Impact of MRD on survival outcomes
The patients who achieved MRD-negative CR at 6 months (n = 16) had significantly longer PFS (not reached vs 17.4 months; P < .0001) and OS (unreached vs 38.2 months; P < .0001) compared with those who did not (n = 4) (Figure 3A-B). At 4 weeks, 9 (50%) of 18 evaluable patients were MRD-free, and MRD at this early time point was also associated with PFS (P = .025) and OS (P = .022). Relevant to patients not followed up by bone marrow testing after treatment, PFS and OS were analyzed with respect to blood MRD at 1 and 6 months after CDAR in all 20 patients. We found that both PFS and OS were significantly longer if blood was MRD negative at either 1 month (P = .0031 for PFS, P = .0017 for OS) or 6 months (P < .0001 for both PFS and OS) (Figure 3C-F).

Survival outcomes by MRD status. Patients with HCLv achieving (n = 16, red) or not achieving (n = 4, blue) MRD-negative CR at 6 months are compared with respect to PFS (A) and OS (B). Similarly, PFS (C, E) and OS (D, F) are shown for patients who were blood MRD-negative (n = 14, red) vs positive (n = 5, blue) at 1 month (C, D), and blood MRD-negative (n = 17, red) vs positive (n = 3, blue) at 6 months (E, F). Patient #11 experienced progression at 4.1 months and is included in the analyses of PFS and OS curves based on results at 6 months because the patient was blood MRD positive at both 1 and 6 months. These 2 sets of analyses could have been done starting at 6 months after beginning treatment to ensure that the trait was considered at the start of the curves but doing so would have eliminated this patient from the PFS curves in panels A and E, and it was judged to be more important to show these full results.
Survival outcomes by MRD status. Patients with HCLv achieving (n = 16, red) or not achieving (n = 4, blue) MRD-negative CR at 6 months are compared with respect to PFS (A) and OS (B). Similarly, PFS (C, E) and OS (D, F) are shown for patients who were blood MRD-negative (n = 14, red) vs positive (n = 5, blue) at 1 month (C, D), and blood MRD-negative (n = 17, red) vs positive (n = 3, blue) at 6 months (E, F). Patient #11 experienced progression at 4.1 months and is included in the analyses of PFS and OS curves based on results at 6 months because the patient was blood MRD positive at both 1 and 6 months. These 2 sets of analyses could have been done starting at 6 months after beginning treatment to ensure that the trait was considered at the start of the curves but doing so would have eliminated this patient from the PFS curves in panels A and E, and it was judged to be more important to show these full results.
Toxicity
Overall, CDAR was well tolerated (Table 3). As expected, because patients with HCLv have higher normal blood counts compared with patients with classic HCL, a relatively low frequency of cytopenias was observed. Only 1 patient each experienced febrile neutropenia and anemia requiring transfusion. As we previously described,6 a transient decrease in platelet count was observed within 24 hours after the first dose of cladribine and rituximab but recovered within days to 1 week. Grade 4 thrombocytopenia occurred in 2 patients, and only 1 received a prophylactic platelet transfusion. Nonhematologic grade 3/4 adverse events were unusual.
Adverse events related to cladribine and rituximab
| Adverse event | Cladribine + rituximab | Delayed rituximab | ||
|---|---|---|---|---|
| (n = 20) | (n = 10) | |||
| All | Grade 3/4 | All | Grade 3/4 | |
| Hematologic | ||||
| Lymphopenia | 12 (60) | 9 (45) | 1 (10) | |
| Leukopenia | 8 (40) | 5 (25) | 1 (10) | |
| Thrombocytopenia | 8 (40) | 6 (30) | ||
| Neutropenia | 4 (20) | 3 (15) | 1 (10) | |
| Febrile neutropenia | 1 (5) | 1 (5) | ||
| Anemia | 1 (5) | 1 (5) | ||
| Microscopic hematuria | 2 (10) | |||
| Low fibrinogen | 1 (5) | |||
| Gastrointestinal and hepatic | ||||
| Elevated transaminases | 7 (35) | |||
| Abdominal/pelvic pain | 1 (5) | |||
| Nausea/vomiting | 2 (10) | |||
| GGT elevation | 2 (10) | |||
| Diarrhea | 2 (10) | |||
| Alkaline phosphatase | 1 (5) | |||
| Anorexia | 1 (5) | |||
| Mucositis | 1 (5) | |||
| Constitutional and pain syndromes | ||||
| Infusion reaction | 11 (55) | |||
| Fatigue | 2 (10) | 1 (10) | ||
| Fever | 2 (10) | |||
| Flu-like symptom | 1 (5) | |||
| Joint pain | 1 (10) | |||
| Diaphoresis | 1 (5) | |||
| Allergic rhinitis | 2 (10) | |||
| Dizziness | 1 (5) | |||
| Immune, infection, and cardiopulmonary | ||||
| Rash/pruritus | 5 (25) | |||
| Pneumonia | 1 (5) | 1 (5) | ||
| Dyspnea/cough | 3 (15) | |||
| Throat pain | 1 (5) | |||
| Other | ||||
| Tumor lysis syndrome | 1 (5) | 1 (5) | ||
| Hypoalbuminemia | 6 (30) | |||
| Edema | 1 (5) | |||
| Hyperuricemia | 1 (5) | 1 (5) | ||
| Hypophosphatemia | 1 (5) | 1 (5) | ||
| Adverse event | Cladribine + rituximab | Delayed rituximab | ||
|---|---|---|---|---|
| (n = 20) | (n = 10) | |||
| All | Grade 3/4 | All | Grade 3/4 | |
| Hematologic | ||||
| Lymphopenia | 12 (60) | 9 (45) | 1 (10) | |
| Leukopenia | 8 (40) | 5 (25) | 1 (10) | |
| Thrombocytopenia | 8 (40) | 6 (30) | ||
| Neutropenia | 4 (20) | 3 (15) | 1 (10) | |
| Febrile neutropenia | 1 (5) | 1 (5) | ||
| Anemia | 1 (5) | 1 (5) | ||
| Microscopic hematuria | 2 (10) | |||
| Low fibrinogen | 1 (5) | |||
| Gastrointestinal and hepatic | ||||
| Elevated transaminases | 7 (35) | |||
| Abdominal/pelvic pain | 1 (5) | |||
| Nausea/vomiting | 2 (10) | |||
| GGT elevation | 2 (10) | |||
| Diarrhea | 2 (10) | |||
| Alkaline phosphatase | 1 (5) | |||
| Anorexia | 1 (5) | |||
| Mucositis | 1 (5) | |||
| Constitutional and pain syndromes | ||||
| Infusion reaction | 11 (55) | |||
| Fatigue | 2 (10) | 1 (10) | ||
| Fever | 2 (10) | |||
| Flu-like symptom | 1 (5) | |||
| Joint pain | 1 (10) | |||
| Diaphoresis | 1 (5) | |||
| Allergic rhinitis | 2 (10) | |||
| Dizziness | 1 (5) | |||
| Immune, infection, and cardiopulmonary | ||||
| Rash/pruritus | 5 (25) | |||
| Pneumonia | 1 (5) | 1 (5) | ||
| Dyspnea/cough | 3 (15) | |||
| Throat pain | 1 (5) | |||
| Other | ||||
| Tumor lysis syndrome | 1 (5) | 1 (5) | ||
| Hypoalbuminemia | 6 (30) | |||
| Edema | 1 (5) | |||
| Hyperuricemia | 1 (5) | 1 (5) | ||
| Hypophosphatemia | 1 (5) | 1 (5) | ||
Data are presented as no. (%) of patients. GGT, gamma-glutamyltransferase.
Discussion
This prospective trial, the largest for HCLv to our knowledge, is unique in following up patients long term for MRD with both bone marrow and blood studies, and the first to show relationships between MRD and survival outcomes. There is no study evaluating purine analogue monotherapy prospectively in HCLv, but of 42 patients with HCLv who received cladribine monotherapy, the CR rate was only 7%.3,7-11 The CR rate of cladribine monotherapy in these 42 patients is far inferior to either the 95% CR rate achieved by CDAR (P < .0001) or the 86% CR rate in 7 patients with HCLv after cladribine followed by rituximab reported by Ravandi et al (P < .0001).15,16 Even if patients respond to cladribine, purine analogue monotherapy does not provide long-term remission; median duration of response was only ∼15 months.3 Despite the comparison of retrospective cladribine monotherapy data vs small prospective trials of cladribine-rituximab, we believe these differences support combination treatment rather than cladribine monotherapy in HCLv.
We found that 80% of 20 patients achieved MRD-negative CR. The most stringent test of MRD was bone marrow aspirate flow cytometry, sensitive to 0.002%. As previously reported by Ravandi et al,15,16 we found this test more sensitive than consensus PCR for immunoglobulin rearrangements in blood or bone marrow. Chihara et al15 reported that 5 (71%) of 7 patients with HCL became MRD negative 3 months after cladribine treatment, which was immediately after completing delayed rituximab therapy, and these patients were not reassessed by bone marrow tests for MRD. In our study, the median duration of MRD-negative CR was 70.1 months, and 7 of 16 remain MRD negative up to 120 months. One-half of the patients have not required any additional treatment, with a median follow-up of 69.7 months and a median potential follow-up of 112.4 months.
MRD status 6 months after CDAR was closely associated with both PFS and OS, and similar importance was found for blood MRD at 1 and 6 months. Although MRD is critical in many other hematologic malignancies,32-35 its clinical value in HCL has been less clear because additional treatment can be delayed so long after MRD recurrence.15,36 However, recent data in HCL show eradication of HCL leading to longer CR duration37 and PFS.38 First-line treatment with CDAR in classic HCL was associated with relapse in 1 of 34 patients at a median follow-up of 6.5 years,17 significantly lower (P = .0023) compared with 28% relapses at 6.5 years in 90 historical patients with baseline cytopenias who had received first-line purine analogue monotherapy.39 Although there is no evidence yet for a benefit in OS for MRD eradication in classic HCL, this study shows for the first time a clear OS as well as PFS benefit for MRD eradication in HCLv, whether assessed by bone marrow or blood only. This is likely due to its more aggressive natural history and lack of alternative therapeutic options. Thus, it is critical to eliminate MRD in effective treatment of HCLv.
TP53 aberrations, including mutation and del(17p), are extremely important in the treatment of malignant disease such as in chronic lymphocytic leukemia and acute myeloid leukemia.40 Loss of function of the TP53 gene is associated with chemoresistance and aggressive disease. The del(17p)/TP53 mutation is rare in HCL but in HCLv affects 30% to 40% of patients.41 We found TP53 mutation in 5 (26%) of 19 evaluable patients tested, and indeed these patients with mutation had shorter response to CDAR with a median PFS of ∼3 years and OS <5 years. This patient population likely represents a significant risk for chemoresistance and suggests unmet needs for non-chemotherapy drug development.
In summary, CDAR has high efficacy in patients with early HCLv, particularly in those achieving MRD-negative CR. However, more than one-half of patients experienced recurrence, and there are still unmet needs, particularly for those not achieving MRD-negative CR or having a TP53 mutation. These findings must be validated in future studies. Although earlier treatment of HCLv with more effective chemoimmunotherapy may hold promise, we observed that some patients with HCLv are or will become chemoresistant.42,43 Therefore, non-chemotherapy approaches, such as moxetumomab pasudotox or ibrutinib42 monotherapy or in combination, should be explored. In this ongoing severe acute respiratory syndrome coronavirus 2 pandemic, these T cell–sparing approaches may be advantageous, but before initiation of a CD20 monoclonal antibody, which can prevent effective immunization for up to 12 months, evidence of adequate levels of COVID-19 spike antibodies may be prudent.44 As always, the choice of therapy needs to be well balanced with eradication of leukemia.45
Acknowledgments
The authors recognize Barbara Debrah for analyzing MRD data; Brian Brown, NIH Library Editing Service, for reviewing the manuscript; research nurses and nurse practitioners involved with the protocol since its inception; nurses and oncology fellows involved in treatment; referring physicians; and the patients and their families. They recognize physicians for often administering delayed rituximab, including Martin Tallman and John Seymour. The authors also thank Maxwell Lee for his valuable discussion on exome-sequencing data analysis and the Hairy Cell Leukemia Foundation for valuable discussions and referrals. This work used the computational resources of the National Institutes of Health HPC Biowulf cluster (http://hpc.nih.gov) and was supported by funding from the Intramural Research Program, National Institutes of Health, National Cancer Institute, Center for Cancer Research.
This study was supported by the intramural program of the National Cancer Institute and GeneTech, Inc.
Authorship
Contribution: R.J.K. designed the study; D.C., S.M.S., C.-H.T., K.P.P., and R.J.K. analyzed the data; E.A. and H.Z. performed whole-exome sequencing; M.S.-S., C.Y., and H.-W.W. performed flow cytometry analysis; M.R., L.X., R.C.B., K.R.C., I.M., and A.D.-F. contributed to the pathological analysis; J.F. and L.J.-E. contributed to patient care; D.C. and R.J.K. wrote the paper; and all authors contributed to finalizing the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Robert J. Kreitman, National Institutes of Health, Building 37/5124b, 9000 Rockville Pike, Bethesda, MD 20892; e-mail: kreitmar@mail.nih.gov.

