

Key Points
This article describes the contribution to immune reconstitution via adoptive cellular therapy for refractory CMV.
These results demonstrate that baseline endogenous immune components correlate with responses in patients treated for refractory CMV.

Abstract
Adoptive cell therapy using cytomegalovirus (CMV)-specific cytotoxic T lymphocytes (CMV-CTLs) has demonstrated efficacy posttransplant. Despite the predicted limited engraftment of CMV-CTLs derived from third-party donors, partially matched third-party donor–derived CMV-CTLs have demonstrated similar response rates to those derived from primary hematopoietic cell transplantation donors. Little is known about the mechanisms through which adoptive cellular therapies mediate durable responses. We performed a retrospective analysis of patients receiving CMV-CTLs for treatment of CMV viremia and/or disease after allogeneic transplant between September of 2009 and January of 2018. We evaluated whether response to adoptively transferred CMV-CTLs correlated with immune reconstitution (IR), using validated CD4+ IR milestones of 50 × 106/L and 200 × 106/L. In this analysis, a cohort of 104 patients received CMV-CTLs derived from a primary transplant donor (n = 25), a third-party donor (n = 76), or both (n = 3). Response to therapy did not increase the likelihood of achieving CD4+ IR milestones at 1 (P = .53 and P > .99) or 2 months (P = .12 and P = .33). The origin of CMV-CTLs did not impact subsequent CD4+ IR. CMV-CTLs appeared to interact with host immunity in mediating responses. Recipients with a baseline CD4 >50 × 106/L had higher response to therapy (P = .02), improved overall survival (P < .001), and protection from CMV-related death (P = .002). Baseline endogenous immunity appears to improve CMV-related and overall survival in this cohort and can be an important marker at the initiation of therapy.
Introduction
Allogeneic hematopoietic cell transplantation (allo-HCT) is a potentially curative treatment for patients with malignant and nonmalignant disorders. Immune reconstitution (IR) starts immediately after allo-HCT with expansion of the innate immune system. Two mechanisms control reconstitution of the adaptive immune system. Initially, there is peripheral expansion of donor T cells or residual host T cells and then de novo T-cell development in the thymus.1,2 The later thymus-dependent development of naive CD4+ T cells can take months to years3 and decreases with age.1
Delayed T-cell reconstitution leads to increased viral infections, transplant-related mortality, all-cause mortality, and relapse.4-7 A previously validated milestone of IR is achievement of CD4 ≥200 × 106/L and phytohemagglutinin mitogen ≥75% of normal.5 Recently, in pediatric cohorts, a newer milestone of early IR, defined as CD4 ≥50 × 106/L by day 100 post–allo-HCT, has been shown to correspond with improved overall survival (OS), as well as decreased transplant-related mortality from viral infections.8-10
Given the importance of T-cell IR after allo-HCT, strategies to hasten IR have been explored, including adoptive transfer of T cells. However, although transfer of T cells can contribute to T-cell immunity (global immunity), it has done so thus far without increasing the incidence of graft-versus-host disease (GvHD) disease.11-13 Adoptive cellular therapy with virus-specific cytotoxic T lymphocytes (CTLs), from an allo-HCT recipient’s primary donor or from banks of third-party (not the hematopoietic cell transplantation [HCT] donor or recipient) donors, has demonstrated efficacy in treating cytomegalovirus (CMV) infections in the post–allo-HCT setting.14-18 Although only 1 study has been able to document the persistence of primary donor–derived virus-specific T cells,19 it is presumed from that study that primary donor viral-specific populations persist long-term. Third-party donor CTLs are presumed to persist only transiently, because expansion of these infused populations has been detected for 90 days.20 Despite this presumed shorter circulation, durable clinical responses can be achieved with third-party donor CTLs.17
We asked whether response to CMV-specific cytotoxic T lymphocytes (CMV-CTLs) correlated with global T-cell IR or whether global T-cell IR could be potentiated by adoptive transfer of CMV-CTLs. Focusing on the immune status of patients prior to and after adoptive cellular therapy, we report the evaluation of a cohort of patients who received CMV-CTLs for refractory CMV infection after allo-HCT and demonstrate that the success of adoptive therapy with CMV-CTLs may rely, in part, on recipient immune components to mediate responses to therapy.
Methods
Study design
We conducted a retrospective analysis of pediatric and adult patients with malignant and nonmalignant disorders who received CMV-CTLs for the treatment of CMV viremia and/or disease after allo-HCT. Patients received primary donor–derived or third-party donor CMV-CTLs at Memorial Sloan Kettering Cancer Center (MSKCC) on 1 of 3 protocols (clinicaltrials.gov NCT02136797, NCT01646645, and NCT00674648) for persistent or refractory CMV. CMV reactivation was defined as a CMV polymerase chain reaction >137 IU/mL on 2 separate occasions, and persistence was defined as active CMV infection or persistent CMV viremia despite treatment with antiviral agents for >2 weeks. Manufacturing of CMV-CTLs occurred as previously described.21 Allo-HCT occurred at MSKCC and collaborating institutions. Data were captured as part of each prospective protocol and retrospective Institutional Review Board (IRB)-approved protocol.
Response, assessed 28 to 42 days after the last dose of each cycle of CMV-CTLs, was defined per protocol as complete response (CR), partial response (PR), stable disease (SD), or progression of disease (POD). Outcomes were categorized as binary, with those achieving CR or PR categorized as responders, whereas those with SD or POD were categorized as nonresponders. Those with additions or changes in concomitant antiviral therapy to treat a non-CMV viral infection, such that the response could not be attributed to CMV-CTLs, were defined as not evaluable. CR was defined as undetectable CMV DNA by polymerase chain reaction and documented clearance of CMV disease in patients with CMV disease at baseline (ie, repeat bronchoalveolar lavage or endoscopy). PR was defined as a 2 × log10 reduction in the level of CMV DNA detected in the blood and, in patients with disease, resolution of clinical symptoms. SD was defined as persistent CMV viremia and, in patients with disease, no change in the clinical severity of disease. POD was defined as disease progression by clinical and radiologic parameters ascribable to CMV infection in any affected organ, with unchanged or increased levels of CMV DNA or initiation of alternative CMV-directed therapy because of clinical concern for progression. Survival information was based on patient status at last follow-up.
Data were captured as part of each prospective protocol and retrospective IRB-approved protocols at MSKCC.
Study end points
Actual cell counts for CD3+, CD4+, and CD8+ T cells, as well as natural killer (NK) and B cells, were collected monthly (± 5 days) from the start of therapy through 6 months on a Clinical Laboratory Improvement Amendments (CLIA)–approved institutional lymphocyte flow cytometry panel (which includes CD3+, CD4+, CD8+, CD19+, CD56+16+, CD45RA+, and CD45+). CD4+ T-cell IR was defined as achieving CD4 ≥ 50 × 106/L and/or CD4 ≥ 200 × 106/L based on validated milestones.5,8,9 Time to IR was defined as the time from first CMV-CTL infusion to the first time that IR was reached. Patients who did not reach IR were censored at the time of the last measure of IR. Given the absence of validated milestones of IR using CD8+ T cells, NK cells, or B cells, analysis of these populations was performed by evaluating the slope of recovery. The slope of recovery was used given interest in evaluation of any change in the pace of immune recovery between recipients of primary donor and third-party donor CMV-CTLs. The slope of recovery of CD4+ T cells, or other cell types, was defined as the log of the level at the start of therapy subtracted from the log of the level after 1 month. Time to response is defined as the time between the first cycle and the day a response was recorded, censoring nonresponders at the time that response was last measured. Non-CMV deaths occurring within 6 months of a nonresponse were considered competing events. Time to CMV death (or death due to other infection) was defined as the time from first CMV-CTL infusion to death from CMV (or other infection, respectively) to be consistent, because time to IR was defined as time from CMV-CTL infusion rather than time from HCT. When analyzing survival by response, the time of origin was the response time for responders. Living patients were censored at their time of last follow-up. Deaths from other causes, including deaths from other infection (or deaths from CMV) were considered competing events in survival analysis.
Statistical analysis
The data cutoff was 10 January 2020. A χ2 test was used to estimate the correlation between response and donor type and between response and baseline immune status. Fisher’s exact test was used for IR at 1, 2, or 3 months. An Aalen-Johansen estimator was used to estimate the cumulative incidence of IR, response, or cause-specific death, as well as the corresponding median times. Differences in cumulative incidences were tested using Gray’s test. The potential follow-up was estimated using a reverse Kaplan-Meier. The association between clinical factors and the probability of achieving CD4 ≥50 × 106/L and/or CD4 ≥200 × 106/L was assessed using a Cox model, in univariable and multivariable models (variables with P < .10 in univariable models were entered in the multivariable model). The impact of CMV-CTL donor type on cell counts (ie, CD4+) was estimated using a longitudinal linear model adjusted for time. Finally, the comparison of the slope of cell counts between responders and nonresponders was estimated using a Mann-Whitney-Wilcoxon test. A P value < .05 was considered statistically significant.
Results
Patient characteristics
Between January of 2008 and January of 2018, 104 pediatric and adult patients received CMV-CTLs for treatment of CMV viremia (n = 74), CMV disease (n = 8), or both (n = 22), arising after allo-HCT. Patient characteristics are shown in Table 1. Three outliers started CMV-CTL therapy at 5.3, 7.6, and 13.5 years from allo-HCT: a patient with Philadelphia-positive B-cell acute lymphoblastic leukemia who had resistant CMV retinitis, another with severe combined immune deficiency who had waning IR after allo-HCT and developed primary CMV infection, and a patient with relapsed chronic lymphoblastic leukemia and chronic GvHD on long-term immune suppression, respectively. At the start of CMV-CTL therapy, 41 patients were on immunosuppressive therapy. Twenty-three of these patients were on steroids, with a median dose of 0.15 mg/kg per day (range, 0.03-0.6 mg/kg per day). Additional immunosuppressive agents included a calcineurin inhibitor (n = 25), mycophenolate (n = 10), sirolimus (n = 5), and/or budesonide (n = 10). Responders (n = 60), nonresponders (n = 25), and nonevaluable patients in whom response to CMV-CTL therapy could not be attributed to CMV-CTLs alone due to changes in antiviral therapy (n = 19) were assessed for survival and cause of death. The 3-year OS was 48% for responders, 12% for nonresponders, and 37% for nonevaluable patients. More than half of the patients in this cohort (n = 67) died during the follow-up, including 20 from CMV disease. Responders were less likely to die from CMV infections than were nonresponders (P < .001). Other causes of death included relapse (n = 14), other infection (n = 15), graft failure (n = 3), GvHD (n = 5), complications of veno-occlusive disease (VOD, n = 1), pulmonary hemorrhage or failure (n = 4), central nervous system hemorrhage (n = 1), autoimmune hemolytic anemia (n = 1), cardiac arrest due to arrhythmia (n = 1), and unknown cause (n = 2). For the entire cohort, the median time to CD4 ≥50 × 106/L was 1.1 months (interquartile range [IQR], 0-3.0), and median time to CD4 ≥200 × 106/L was 4.4 months (IQR, 2.1-7.5).
Patient characteristics
| Patient characteristics | Primary donor (n = 25) | Third party (n = 76) | Both (n = 3) | All (N = 104) |
|---|---|---|---|---|
| Age, median (range), y | 55.9 (0.6-73.0) | 49.9 (0.3-69.7) | 62.0 (13.4-67.4) | 51.8 (0.3-73.0) |
| Sex, male/female, n | 12/13 | 40/36 | 1/2 | 53/51 |
| Malignant/nonmalignant, n | 21/4 | 64/12 | 2/1 | 87/17 |
| Stem cell source, n | ||||
| Conventional BM | 1 | 5 | 1 | 7 |
| Conventional PBSC | 1 | 12 | 0 | 13 |
| Unrelated cord | 0 | 9 | 0 | 9 |
| CD34-selected TCD PBSC | 22 | 48 | 2 | 72 |
| CD34-selected TCD BM | 1 | 1 | 0 | 2 |
| PT Cy TCD | 0 | 1 | 0 | 1 |
| CMV serostatus, n | ||||
| D+/R+ | 24 | 29 | 3 | 56 |
| D−/R+ | 0 | 38 | 0 | 38 |
| Unknown | 1 | 9 | 0 | 10 |
| HLA matching, n | ||||
| Matched | 19 | 32 | 3 | 54 |
| Mismatched | 6 | 44 | 0 | 50 |
| HCT to CMV reactivation (range), d | 28 (4-2520) | 27 (38-4655) | 24 (19-82) | 28 (38-4655) |
| HCT to first CTLs (range), d | 116 (76-2763) | 132 (29-4940) | 128 (59-385) | 128 (29-4940) |
| Immunosuppression at first CTLs, n | 4 | 36 | 1 | 41 |
| Number of cycles, n | ||||
| 1 | 21 | 40 | 0 | 61 |
| 2 | 2 | 22 | 0 | 24 |
| 3 | 1 | 6 | 3 | 10 |
| ≥4 | 1 | 8 | 0 | 9 |
| Response, n | ||||
| Responder (CR/PR) | 17 | 40 | 3 | 60 |
| Nonresponder (SD/POD) | 4 | 21 | 0 | 25 |
| Not evaluable | 4 | 15 | 0 | 19 |
| Death from CMV, n | 7 | 12 | 1 | 20 |
| Patient characteristics | Primary donor (n = 25) | Third party (n = 76) | Both (n = 3) | All (N = 104) |
|---|---|---|---|---|
| Age, median (range), y | 55.9 (0.6-73.0) | 49.9 (0.3-69.7) | 62.0 (13.4-67.4) | 51.8 (0.3-73.0) |
| Sex, male/female, n | 12/13 | 40/36 | 1/2 | 53/51 |
| Malignant/nonmalignant, n | 21/4 | 64/12 | 2/1 | 87/17 |
| Stem cell source, n | ||||
| Conventional BM | 1 | 5 | 1 | 7 |
| Conventional PBSC | 1 | 12 | 0 | 13 |
| Unrelated cord | 0 | 9 | 0 | 9 |
| CD34-selected TCD PBSC | 22 | 48 | 2 | 72 |
| CD34-selected TCD BM | 1 | 1 | 0 | 2 |
| PT Cy TCD | 0 | 1 | 0 | 1 |
| CMV serostatus, n | ||||
| D+/R+ | 24 | 29 | 3 | 56 |
| D−/R+ | 0 | 38 | 0 | 38 |
| Unknown | 1 | 9 | 0 | 10 |
| HLA matching, n | ||||
| Matched | 19 | 32 | 3 | 54 |
| Mismatched | 6 | 44 | 0 | 50 |
| HCT to CMV reactivation (range), d | 28 (4-2520) | 27 (38-4655) | 24 (19-82) | 28 (38-4655) |
| HCT to first CTLs (range), d | 116 (76-2763) | 132 (29-4940) | 128 (59-385) | 128 (29-4940) |
| Immunosuppression at first CTLs, n | 4 | 36 | 1 | 41 |
| Number of cycles, n | ||||
| 1 | 21 | 40 | 0 | 61 |
| 2 | 2 | 22 | 0 | 24 |
| 3 | 1 | 6 | 3 | 10 |
| ≥4 | 1 | 8 | 0 | 9 |
| Response, n | ||||
| Responder (CR/PR) | 17 | 40 | 3 | 60 |
| Nonresponder (SD/POD) | 4 | 21 | 0 | 25 |
| Not evaluable | 4 | 15 | 0 | 19 |
| Death from CMV, n | 7 | 12 | 1 | 20 |
BM, bone marrow; D+, donor positive; D−, donor negative; PBSC, peripheral blood stem cell; PT Cy, posttransplant cyclophosphamide; R+, recipient positive; TCD, T-cell depleted.
CD4+ T-cell IR after the start of treatment with CMV-CTLs
Eighty-five patients had known response statuses attributable to CMV-CTL therapy and were included in the analysis of IR. Overall, 71% (60/85) were categorized as responders and 29% (25/85) were nonresponders to therapy. Of evaluable patients with a baseline CD4 <50 × 106/L (n = 52), 27 achieved a CD4 ≥50 × 106/L at some point during therapy. Achievement of this immune milestone was incremental over the course of adoptive cell therapy. At 1 month after initiation of therapy, only 2 of the 32 responding patients (6%) achieved a CD4 ≥50 × 106/L, whereas none of the 18 nonresponders achieved this milestone (P = .53). At 2 months after initiation of therapy, 41% (13/32) of the responders had achieved a CD4 ≥50 × 106/L compared with 17% (3/18) of the nonresponders (P = .12). At 3 months after initiation of therapy, 62% (20/32) of the responders had achieved a CD4 ≥50 × 106/L compared with 33% (6/18) of the nonresponders (P = .08; Figure 1A).
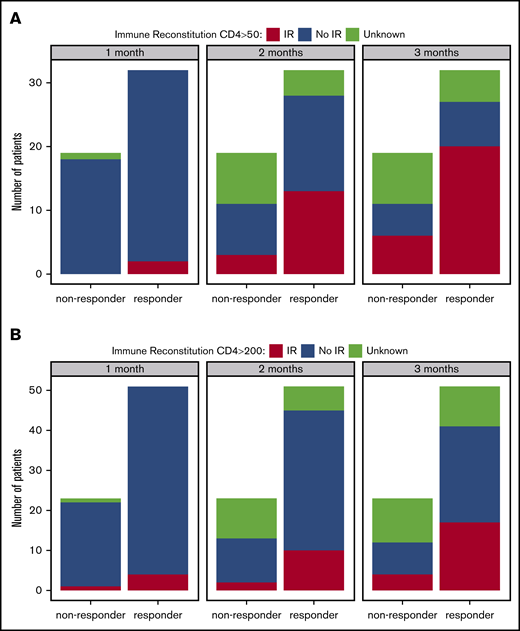
Response did not affect the achievement of either IR milestone at select time points. (A) Percentage of patients who achieved IR (CD4 ≥50 × 106/L) at different time points according to response (1-month P = .53; 2-month P = .12; 3-month P = .08). (B) Percentage of patients who achieved IR (CD4 ≥200 × 106/L) at different time points according to response (1-month P > .99; 2-month P = .33; 3-month P = .26).
Response did not affect the achievement of either IR milestone at select time points. (A) Percentage of patients who achieved IR (CD4 ≥50 × 106/L) at different time points according to response (1-month P = .53; 2-month P = .12; 3-month P = .08). (B) Percentage of patients who achieved IR (CD4 ≥200 × 106/L) at different time points according to response (1-month P > .99; 2-month P = .33; 3-month P = .26).
In addition, of 74 evaluable patients with a baseline CD4 <200 × 106/L, 34 achieved a CD4 ≥200 × 106/L at some point during therapy. Among those, 26 (76%) were responders, whereas 8 (24%) were nonresponders. At 1 month after initiation of therapy, 8% (4/51) of responding patients achieved a CD4 ≥200 × 106/L compared with 5% (1/22) of the nonresponders (P > .99). At 2 months after initiation of therapy, 20% (10/51) of responders achieved a CD4 ≥200 × 106/L compared with 9% (2/22) of the nonresponders (P = .33). Finally, at 3 months after initiation of therapy, 33% (17/51) of the responders achieved a CD4 ≥200 × 106/L compared with 18% (4/22) of the nonresponders (P = .26; Figure 1B). There was no evidence that responders to therapy were more likely to achieve an IR milestone of CD4 ≥ 50 × 106/L or ≥200 × 106/L than were nonresponders.
To assess the kinetics of T-cell reconstitution, we evaluated the log of slope of CD4+ and CD8+ T cells and found there was no difference between the slope of absolute CD4+ T-cell numbers in responders (mean = 1.09) and the slope in nonresponders (mean = 0.86; P = .69). Similarly, there was no difference in the slope of absolute CD8+ T-cell numbers between responders (mean = 1.17) and nonresponders (mean = 1.48; P = .57).
Univariable and multivariable analysis identified factors prognostic of IR. In multivariable analysis, the chance of reaching CD4 ≥50 × 106/L during follow-up was increased for patients receiving an unmodified transplant (conventional BM, PBSC and cord blood) compared with a T-cell–depleted transplant (HR, 2.26; 95% CI, 1.35-3.78; P = .002), and increased by 5% for every 100 days elapsed between transplant and the first CTL cycle (HR, 1.05; 95% CI, 1.02-1.08, P = .001). Immune suppression, age at allo-HCT, and HLA matching were not associated with the chance of reaching CD4 ≥50 × 106/L (P = .15, .17, and >.99 respectively). In addition, the chance of achieving CD4 ≥200 × 106/L during follow-up was decreased for older patients, by 8% for every 5-year difference (hazard ratio [HR], 0.92; 95% CI, 0.86-0.99; P = .02), whereas the chance of achieving CD4 >200 × 106/L was not significantly associated with immune suppression (P = .59), type of transplant (T-cell depleted vs others; P = .61), HLA matching (P = .71), or the time from allo-HCT to first CTL (P = .07).
Baseline CD4 IR prior to the start of therapy
A baseline CD4 < 50 × 106/L was seen in 63% (52/83) of evaluable patients and was associated with a failure to respond. Patients with a baseline CD4 ≥ 50 × 106/L had a higher chance of responding than did those with a baseline CD4 < 50 × 106/L (P = .02; Figure 2). The 4-month CI of response was 88% (95% confidence interval [CI]: 76-100) in patients with a baseline CD4 ≥ 50 × 106/L compared with 71% (95% CI, 56-85) in those with a baseline CD4 < 50 × 106/L. An increased OS was seen in patients with a baseline CD4 ≥ 50 × 106/L compared with those with a baseline CD4 < 50 × 106/L, with respective 3-year rates of 63.5% (95% CI, 47.6-79.4) and 22.1% (95% CI, 11.5-32.7) (P < .001). Twelve of 21 nonresponding patients with a baseline CD4 < 50 × 106/L died from CMV compared with 0 of 4 nonresponding patients with a baseline CD4 ≥ 50 × 106/L. The 12-month risk of death from CMV was lower in patients with a baseline CD4 ≥ 50 × 106/L (0%) compared with patients with a baseline CD4 < 50 × 106/L (29%; 95% CI, 17-40), demonstrating the prognostic value of baseline CD4 count in death from CMV (P = .002).

Response to CMV-CTLs differs according to baseline CD4+counts. Cumulative rate of response to CMV-CTLs between baseline CD4 ≥50 × 106/L and baseline CD4 <50 × 106/L (P = .02).
Response to CMV-CTLs differs according to baseline CD4+counts. Cumulative rate of response to CMV-CTLs between baseline CD4 ≥50 × 106/L and baseline CD4 <50 × 106/L (P = .02).
Donor type impact on IR
Response rates did not differ between recipients of primary donor and third-party donor CMV-CTLs (81% [17/21] and 66% [40/61], respectively; P = .30). A longitudinal analysis looking at the log of CD4+ and CD8+ over time did not find any significant difference between recipients of primary donor–derived and third-party donor–derived CMV-CTLs (Figure 3A, P = .28; Figure 3B, P = .13, respectively). The 3-month cumulative rate of CD4 ≥50 × 106/L was 71% (95% CI, 40-100) in the primary donor–derived cohort and 66% (95% CI, 49-83) in the third-party cohort. The donor type did not have any significant impact on the number of CD3+ T cells (P = .15) or B cells (P = .45). Recipients of third-party donor CMV-CTLs had a higher level of NK cells on average: log-NK cells were higher by 0.55 cells/μL (95% CI, 0.02-1.08) (P = .04).

The log of CD4+and CD8+over time does not differ between donor types. (A) Mean and 95% CI for log of CD4+ over time by donor type. The dashed line represents the CD4 = 200 × 106/L threshold (P = .28). (B) Mean and 95% CI for log of CD8+ over time by donor type (P = .13).
The log of CD4+and CD8+over time does not differ between donor types. (A) Mean and 95% CI for log of CD4+ over time by donor type. The dashed line represents the CD4 = 200 × 106/L threshold (P = .28). (B) Mean and 95% CI for log of CD8+ over time by donor type (P = .13).
Discussion
In HCT recipients, refractory CMV infections manifested by viremia and/or disease are associated with high nonrelapse mortality.22 Adoptive cellular therapy is an increasingly available modality to treat CMV infections refractory to standard antiviral therapy.17,23,24 Durable responses to this approach have been demonstrated with at least transient expansion of CMV-specific T cells and CD8+ T cells.24,25 Recently, T-cell repertoire remodeling26 and emergence of an immune signature of CMV control have been identified in recipients of CMV-CTLs who achieve control.27 Recipient immunity as a determinant of response has not yet been assessed. To assess whether the durability of response relates to baseline recipient immunity or improvement in IR mediated by adoptively transferred T cells, we evaluated a cohort of patients who had frequent immune monitoring performed prior to, during, and after adoptive transfer of CMV-specific T cells. This report demonstrates that endogenous immunity may play a role in response to therapy.
Multiple factors impact IR posttransplant. CMV and IR have been shown to be bidirectional. The deficiency in endogenous T cells posttransplant allows for the proliferation of CMV, and they are required for control and treatment of the virus.28 However, it has been demonstrated that CMV reactivation can increase the number of CMV antigen–specific T cells, and some studies have even suggested delaying treatment if possible to allow for low-level reactivation.29,30 Because CMV can also be responsible for graft failure and delayed IR,28,31 other investigators recommend close monitoring and rapid initiation of antiviral therapy at the first indication of reactivation.32 In addition, treatment with ganciclovir and maribavir has been shown to further delay IR.33,34 In the current study, patients received CMV-CTLs at a median of 128 days after transplant and had CMV that was refractory to multiple lines of antiviral therapy. This likely is a result of and contributes to their poor CMV-specific and overall IR, helping to explain the overall dismal survival of the nonresponding patients.
It should be emphasized that IR (CD4 ≥50 × 106/L and CD4 ≥200 × 106/L) previously has been assessed from the time of transplant, whereas we evaluated it from the time of initiation of adoptive T-cell therapy. Although response and achievement of IR increased during the evaluation period, we were unable to demonstrate that response to therapy correlated with the achievement of a CD4 ≥50 × 106/L (in patients who started with minimal endogenous immunity) or CD4 ≥200 × 106/L. Rather, time from transplant, T-cell depletion vs conventional transplant, and age were found to predict IR, as they were previously shown to do when evaluated in the first 100 days post–allo-HCT. Although it was disappointing that adoptively transferred CMV-CTLs did not appear to mediate more global IR, this analysis was limited by the increased mortality in nonresponders that made it difficult to fully compare IR in responding vs nonresponding recipients over time. Thus, it remains possible that global IR is potentiated by adoptively transferred virus-specific T cells. This analysis could be performed in recipients of CMV-CTLs earlier in their course or in recipients of adoptive T-cell therapy given for a viral infection, such as BK virus, not associated with increased mortality.
We used an IR milestone of a CD4 ≥50 × 106/L to define a level of minimal IR based on the prognostic value of CD4+ IR in the first 100 days after transplant and protection against reactivation of some viruses, such as adenovirus, but not CMV.8-10 We found that patients with a baseline CD4 ≥50 × 106/L before starting therapy had a higher cumulative rate of response to therapy over time and a superior OS rate compared with those whose levels were below this threshold. We also evaluated the prognostic value of this milestone in CMV-related death and found that a baseline CD4 ≥50 × 106/L was protective in responding and nonresponding patients.
Comparisons of primary donor and third-party donor recipients did not reveal any differences in percentages of response, slopes of CD4+ and CD8+ T-cell recovery over time, or achievement of IR milestones, despite differences in persistence. In most studies, third-party virus-specific T cells do not appear to durably engraft, but they still mediate durable responses.35 There are ≥3 possible explanations: they engraft at low levels beyond the threshold of detection and provide long-term protection, they simply provide a bridge until the endogenous virus-specific T cells recover, and/or they help to mediate recovery of the endogenous virus-specific T-cell responses. The results of our analysis support the idea that the recipient’s immune system is already “poised” to respond or that an immune interaction occurs between adoptively transferred and recipient T cells. We speculate that third-party T cells could attract endogenous T cells to sites of infection by recognition of the allogeneic T cells and/or by increasing local antigen presentation. This level of speculation is beyond that supported by the present data and study.
In summary, adoptive therapy with CMV-CTLs may rely on recipient immune components to mediate durable responses to therapy and protect patients from CMV-related death. Although the products infused are not clones and contain CD4+ and CD8+ T cells, adoptive T-cell therapy for CMV is focused on delivering T cells with CMV-directed cytotoxicity. It has been demonstrated that CD4+ is critical for durable control of CMV,36 and this study may help to identify a threshold of recipient CD4+ T cells in that role. Limitations of this study include its retrospective nature and the inability to compare responding and nonresponding patients in terms of long-term immunity as a result of the high number of early patient deaths. The incidence of CMV-related deaths, especially in the nonresponding patients, underscores the high-risk nature of this population. Future directions should include expanded phenotyping of the infused T cells, as well as those prior to and after therapy, in the recipients to determine whether allogeneic adoptive cellular therapy changes the T-cell repertoire, as seen with autologous therapy.26 These data provide a platform for incorporating baseline endogenous immunity and IR milestones into the design of multicenter prospective studies using adoptive cellular therapy to treat viral infections after allo-HCT.
Data sharing requests should be sent to Susan E. Prockop (prockops@mskcc.org).
Acknowledgments
The authors thank the patients and families who were treated on these clinical trials and Joseph Olechnowicz (Department of Pediatrics, MSKCC) for editorial assistance.
This work was unfunded, but it was supported in part by National Institutes of Health/National Center for Advancing Translational Sciences grant UL1-TR-002384 and National Institutes of Health/National Cancer Institute Cancer Center Support Grant P30CA008748. Atara Biotherapeutics previously provided support for clinical trials NCT02136797 and NCT01646645. S.E.P. received research support through MSKCC for the conduct of sponsored trials (NCT02136797, NCT01646645, and NCT00674648). Atara Biotherapeutics did not support, modify the study design, or participate in the analysis or interpretation of data for this article. Data were captured as part of each prospective protocol and retrospective IRB-approved protocols.
Authorship
Contribution: V.A.F., A.M., and S.E.P. analyzed the data and created the figures; V.A.F. and S.E.P. wrote the manuscript; and all authors designed the research, performed research (treated patients/collected data), and critically reviewed and approved the content of the manuscript.
Conflict-of-interest disclosure: R.J.O. and E.D. have received consulting fees and grant support from Atara Biotherapeutics, the Starr Cancer Consortium, and National Cancer Institute P01 CA23766. When Atara Biotherapeutics licensed banks of Epstein-Barr virus and CMV T cells, R.J.O. and E.D. received part of the royalties paid to MSKCC. MSKCC holds several patents related to this work on which R.J.O. and E.D. are inventors. S.E.P. receives support for the conduct of sponsored trials from Atara Biotherapeutics, Mesoblast, and Jasper. S.E.P. is an inventor of intellectual property licensed to Atara Biotherapeutics by MSKCC; S.E.P. assigned all rights to MSKCC and has no financial interest in Atara Biotherapeutics. The remaining authors declare no competing financial interests.
Correspondence: Susan E. Prockop, Memorial Sloan Kettering Cancer Center, 1275 York Ave, New York, NY 10065; e-mail: prockops@mskcc.org.

