

Key Points
ClinGen PD-EP adapted ACMG/AMP variant curation and interpretation criteria for ITGA2B and ITGB3, genes underlying autosomal recessive GT.
Adapted criteria were further refined through validation using 70 pilot ITGA2B/ITGB3 variants to produce final GT-specific criteria.

Abstract
Accurate and consistent sequence variant interpretation is critical to the correct diagnosis and appropriate clinical management and counseling of patients with inherited genetic disorders. To minimize discrepancies in variant curation and classification among different clinical laboratories, the American College of Medical Genetics and Genomics (ACMG), along with the Association for Molecular Pathology (AMP), published standards and guidelines for the interpretation of sequence variants in 2015. Because the rules are not universally applicable to different genes or disorders, the Clinical Genome Resource (ClinGen) Platelet Disorder Expert Panel (PD-EP) has been tasked to make ACMG/AMP rule specifications for inherited platelet disorders. ITGA2B and ITGB3, the genes underlying autosomal recessive Glanzmann thrombasthenia (GT), were selected as the pilot genes for specification. Eight types of evidence covering clinical phenotype, functional data, and computational/population data were evaluated in the context of GT by the ClinGen PD-EP. The preliminary specifications were validated with 70 pilot ITGA2B/ITGB3 variants and further refined. In the final adapted criteria, gene- or disease-based specifications were made to 16 rules, including 7 with adjustable strength; no modification was made to 5 rules; and 7 rules were deemed not applicable to GT. Employing the GT-specific ACMG/AMP criteria to the pilot variants resulted in a reduction of variants classified with unknown significance from 29% to 20%. The overall concordance with the initial expert assertions was 71%. These adapted criteria will serve as guidelines for GT-related variant interpretation to increase specificity and consistency across laboratories and allow for better clinical integration of genetic knowledge into patient care.
Introduction
With the rapid advancements in sequencing technology in the last 2 decades, most molecular diagnostic laboratories have adopted next-generation sequencing (NGS) in clinical genetic testing of inherited disorders. The broad genomic coverage and high-throughput nature of NGS-based tests have led to accelerated discoveries of new disease-causing genes and variants. An enormous number of novel sequence variants continue to be identified every day. Accurate interpretation of sequence variants is of critical importance in the diagnosis, management, and genetic counseling of patients with inherited disorders. Interlaboratory discrepancies in variant classification methodologies and the high complexity of the human genome were recognized as barriers to consistent and accurate variant interpretation.1-3 To address this challenge, the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) jointly made recommendations for the interpretation of sequence variants in 2015.4 This guideline report described a process for classifying sequence variants into 5 interpretative categories and recommended the use of a standard terminology system: pathogenic, likely pathogenic, uncertain significance, likely benign, and benign for variants identified in Mendelian disorders. The guideline further recommended all assertions of pathogenicity to be reported with respect to a condition and inheritance pattern. The variant classification is based on evidence including population data, computational data, functional data, and segregation data. Two sets of interpretation criteria were provided: 1 for pathogenic (P) evidence, and 1 for benign (B) evidence. Each pathogenic criterion was weighted as very strong (PVS1), strong (PS1-4), moderate (PM1-6), or supporting (PP1-5). Each benign criterion was weighted as standalone (BA1), strong (BS1-4), or supporting (BP1-6). The classification of variants of clinical significance is based on the combination of fulfilled criteria. If a variant does not fulfill criteria for clinical significance, or if the evidence for benign and pathogenic classification is conflicting, the variant is classified as a variant of uncertain significance (VUS). This set of consensus recommendations advanced the field by defining standardized criteria as well as categories of variant classification, reducing the degrees of subjectivity and variability in variant interpretation.
To centralize knowledge and resources, ClinVar and the Clinical Genome Resource (ClinGen) serve as a central database for clinically classified variants and as a body for managing clinically relevant genomic knowledge, respectively.5 The landmark ACMG/AMP guideline was designed to be universally applicable; however, the categorization of variants using the guideline rules still showed interlaboratory variation. Optimal application of the general guidelines to specific diseases and genes is enhanced by appropriate adaptation of rules informed by clinical domain expertise. To meet this need, ClinGen initiated the formation of Variant Curation Expert Panels (EPs) to adapt the ACMG/AMP criteria to specific genes or disorders.6 By standardizing gene-specific variant curation guidelines, sharing data among expert members, and incorporating data from existing disease databases, the working group aims to decrease the number of VUSs and clarify prior designations based on insufficient evidence, thereby improving the value of genetic testing as a clinical diagnostic tool. The first Variant Curation EP in the clinical domain of hemostasis/thrombosis, the Platelet Disorder EP (PD-EP), was supported by the American Society of Hematology (ASH) and tasked to establish ACMG/AMP rule specifications for interpretation of variants resulting in inherited platelet disorders (IPDs).
Inherited platelet disorders are a heterogeneous group of disorders characterized by abnormal platelet function and/or number, with variable bleeding.7,8 The expression of many IPD genes is not restricted to platelets or the hematopoietic system; therefore, some patients with IPDs present with syndromic features. Although the well-characterized clinical and laboratory phenotypes of the major platelet function disorders often enable the correct diagnosis to be reached, for the more prevalent and often phenotypically milder IPDs, diagnosis can be challenging.8 Genetic testing is of great clinical value in accurately identifying or efficiently confirming the diagnosis, in turn informing optimal treatment, predicting disease progression, and providing genetic counseling for the patients and their families.8,9
ITGA2B and ITGB3, which underlie the prototypic platelet function disorder Glanzmann thrombasthenia (GT), were the initial genes selected by the PD-EP to develop variant interpretation rule specifications, which will inform rule specifications for other IPDs. GT is a disorder of platelet function, inherited in an autosomal recessive pattern, resulting from quantitative or qualitative defects in integrins αIIb and β3 (forming platelet glycoprotein IIb/IIIa) and caused by homozygous or compound heterozygous pathogenic genetic alterations in either ITGA2B or ITGB3.9 Most patients with GT present with severe mucocutaneous bleeding at an early age, a specific pattern of abnormal platelet aggregation, and absent to reduced platelet integrin αIIbβ3 expression, but normal platelet count, size, and granularity. GT is divided into 3 types: type 1 with absent αIIbβ3 expression (<5% of normal), type 2 with reduced αIIbβ3 expression (5% to 25% of normal), and type 3 with normal levels (50% to 100%) but nonfunctional or dysfunctional αIIbβ3. More recently, ITGA2B/ITGB3-related macrothrombocytopenia has been described, in which pathogenic variants of these genes cause spontaneous activation of αIIbβ3 and interfere with proplatelet formation, resulting in an autosomal dominant mild to moderate thrombocytopenia with absent/mild bleeding.9-12 Rule specifications for this IPD phenotype will be considered separately by the PD-EP.
Combining all ITGA2B/ITGB3 variants identified in the literature, the Glanzmann Thrombasthenia Database,13 and ClinVar, the PD-EP had identified a total of 719 variants (388 ITGA2B variants and 331 ITGB3 variants) as of 13 October 2020. The composition of variant types is: 61% single-nucleotide variants within coding regions, 20% insertion/deletions, 11% canonical splice site or deeper intronic variants, and 8% untranslated region variants. The prior reports on GT variants showed extreme heterogeneity of genetic and physiological characterizations that dampened confidence regarding claims of pathogenic variants. The PD-EP, consisting of 21 professionals from 14 institutions and 5 countries with expertise in diagnosis and treatment of platelet disorders, laboratory assays for platelet function testing, inherited platelet disorder research, and molecular genetic diagnostics and variant classification, developed ACMG/AMP rule specifications for interpreting ITGA2B and ITGB3 variants and then optimized the rules following pilot classifications. We herein report the GT-specific sequence variant classification rules, the rationales for each modification of original ACMG/AMP criteria, and the results of the pilot variant curation.
Methods
ClinGen PD-EP
In 2017, the ClinGen Hemostasis/Thrombosis Clinical Domain Working Group assembled an executive committee of clinicians and researchers with expertise in hemostasis and thrombosis who prioritized the curation of genes and variants within several relevant domains. EPs were developed to address both gene curation (assessing the strength of association between a particular gene and a disease phenotype) and variant curation (assessing the pathogenicity of variants within selected genes with respect to a specific phenotype). The pioneer Hemostasis/Thrombosis EP was launched in 2018 when the PD-EP was organized with support from ASH.
All PD-EP members disclosed potential conflicts of interest as required by ClinGen. Approval of variant curation EPs is overseen by ClinGen and consists of 4 steps: (1) defining the group/members and scope of work, (2) developing gene/disease-specific classification rules, (3) optimizing rules using pilot variants, and (4) implementing specified variant curation in the ClinGen Variant Curation Interface14 with submission of curated variants to the ClinVar database.15
ACMG/AMP specifications for ITGA2B/ITGB3
Rule specification for ITGA2B and ITGB3 with respect to GT was accomplished via monthly teleconferences and by convening at the ASH Annual Meetings. Three subgroups were developed to streamline the specification process: clinical phenotype, functional, and computational/predictive/population data. Suggestions for specification of each subgroup of criteria were gathered from all PD-EP members by e-mail, followed by teleconference discussion to reach consensus. PD-EP members proposed and discussed changes to the existing ACMG/AMP criteria, including disease- or gene-specific modifications, strength-level adjustments, and judgment of criteria not applicable to ITGA2B/ITGB3 or GT. Recommendations from the ClinGen Sequence Variant Interpretation (SVI) Working Group were also incorporated.16
Pilot variant classification and specification refinement
The ITGA2B/ITGB3 GT-related specifications were validated with 70 pilot variants, representing 35 each for ITGA2B and ITGB3. Variants were nominated by PD-EP members selecting from ClinVar, the Glanzmann Thrombasthenia Database,13 and internal laboratory data, with the objective of including a balance of known/suspected pathogenic or benign variants (67%) and VUSs or variants with conflicting assertions (33%; Figure 1). Additionally, variants were selected to represent various types of alterations: missense (56%), nonsense and frameshift (14%), splicing (9%), inframe indel (3%), intronic (10%), and synonymous (9%). All pilot variants are annotated using RefSeq IDs NM_000419.4 (ITGA2B), NM_000212.2 (ITGB3), and NC_000017.11 (GRCh38/hg38).

Schematic representation of integrin αIIbβ3 major domains in relation to 70 pilot variants with their final PD-EP classification. Integrin αIIbβ3 subunits αIIb (top) and β3 (bottom) each possess a signal peptide (SP), an extracellular domain involved in ligand binding (β propeller and βI, respectively), a transmembrane (TM) domain, and a cytoplasmic domain (C). Clinically actionable variants (PATH, LPATH) are shown above the protein schematics; VUS, LBEN, and BEN variants are shown below.
Schematic representation of integrin αIIbβ3 major domains in relation to 70 pilot variants with their final PD-EP classification. Integrin αIIbβ3 subunits αIIb (top) and β3 (bottom) each possess a signal peptide (SP), an extracellular domain involved in ligand binding (β propeller and βI, respectively), a transmembrane (TM) domain, and a cytoplasmic domain (C). Clinically actionable variants (PATH, LPATH) are shown above the protein schematics; VUS, LBEN, and BEN variants are shown below.
Each variant was assessed by a curator trained to perform variant interpretation using the 2015 ACMG/AMP guidelines and the PD-EP specifications. The curator selected the relevant criteria based on the evidence provided by PD-EP members and data retrieved from published literature and databases. Curators used the ClinGen Variant Curation Interface to document the applicable criteria and evidence for each variant. Evidence used for interpretation was presented to the full PD-EP for review and final classification based on group consensus. Preliminary guideline specifications were refined throughout the pilot curation process, with variants illustrative of debated criteria used to facilitate additional guideline refinements. Upon ClinGen step-4 approval, the classified ITGA2B/ITGB3 variants were submitted to ClinVar under a 3-star (expert panel–reviewed) status as part of the US Food and Drug Administration–recognized database. The first 115 interpretations are now available in ClinVar and can be accessed online.17
Results
ACMG/AMP criteria specification for ITGA2B/ITGB3
The PD-EP specifications to the ACMG/AMP variant curation criteria for ITGA2B and ITGB3 in relation to GT are summarized in Table 1. Gene- or disease-based specifications were made to 16 ACMG/AMP criteria, including 7 specifying modifications of rule strength based on the quantity or quality of available evidence. Another 7 criteria were deemed not applicable because of lack of relevance to ITGA2B/ITGB3 or GT, and the remaining 5 criteria were determined to be applicable as originally described without further specification. One modification was made to the rules outlined by ACMG/AMP for combining criteria to arrive at a classification: a likely pathogenic classification can be reached if a variant satisfies 1 very strong criterion and 1 supporting criterion. Although not included in the original guideline, this combination of 1 very strong criterion and 1 supporting criterion is consistent with the posterior probability >0.90 for likely pathogenic as described in the Bayesian model of the classification framework.18
PD-EP specifications of the ACMG/AMP guidelines for ITGA2B/ITGB3 variants in relation to GT
| ACMG/AMP criteria | Original ACMG/AMP criteria summary | Specification | Standalone | Very strong | Strong | Moderate | Supporting | Comments |
|---|---|---|---|---|---|---|---|---|
| PVS1 | Null variant in a gene where LOF is a known mechanism of disease | Gene specific | N/A | Per modified PVS1 decision tree (Figure 2) with specified regions critical to protein function | N/A | Three critical regions: (1) the cytoplasmic domain of the β3 subunit, (2) the transmembrane domains of αIIbβ3, and (3) the extracellular domains of αIIbβ3 involved in ligand binding | ||
| PS1 | Same amino acid change as a previously established pathogenic variant regardless of nucleotide change | None | N/A | N/A | Use per original guidelines | N/A | N/A | Both variants must be classified using ITGA2B/ITGB3-specified guidelines |
| PS2 | De novo in a patient (maternity and paternity confirmed) with the disease and no family history | Disease specific, strength | N/A | Use the SVI-recommended point system (supplemental Table 1) based on confirmed vs assumed maternity/paternity status, the number of de novo probands, and the phenotypic consistency | (1) To be scored at phenotype highly specific for gene, the patient must meet the PP4 criteria; (2) proband must harbor an additional pathogenic or likely pathogenic variant with the de novo variant | |||
| PS3 | Well-established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product | Gene specific, strength | N/A | N/A | <5% surface expression of αIIbβ3 (measured by flow cytometry or western blot) and/or significantly reduced function (ie, minimal binding to fibrinogen or ligand mimetic antibodies) | 5%-25% surface expression of αIIbβ3 (measured by flow cytometry or western blot) | N/A | All assays must be performed in a heterologous cell line or animal model with expression of the variant of interest from 1 gene (eg, ITGA2B) and the wild type of the opposite gene (eg, ITGB3) |
| PS4 | The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls | N/A | Rule does not apply because of the rarity of disorder and lack of appropriate studies | |||||
| PM1 | Located in a mutational hotspot and/or critical and well-established functional domain without benign variation | N/A | Rule does not apply because of highly polymorphic nature of genes and no known hotspots | |||||
| PM2 | Absent in population databases (or at extremely low frequency if recessive) | Disease specific | N/A | N/A | N/A | N/A | <1 in 10 000 (0.0001) alleles in all gnomAD continental population cohorts; not present in homozygous state | |
| PM3 | For recessive disorders, detected in trans with a pathogenic variant | Disease-specific, Strength | N/A | Use SVI point recommendations (supplemental Table 2); strength may be adjusted based upon confirmation of variant phasing and classification of the second variant | Both variants must be below the PM2 threshold, and both must be classified using ITGA2B/ITGB3-specified guidelines | |||
| PM4 | Protein length changes as a result of inframe deletions/insertions in a nonrepeat region or stop-loss variants | None | N/A | N/A | N/A | Use per original guidelines | N/A | |
| PM5 | Novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before | Strength | N/A | N/A | N/A | Use per original guidelines | Novel missense change at an amino acid residue where a different missense change determined to be likely pathogenic has been seen before | Both variants must be classified using ITGA2B/ITGB3-specified guidelines |
| PM6 | De novo in a patient (maternity and paternity not confirmed) with the disease and no family history | Disease specific, strength | N/A | Use the SVI-recommended point system (supplemental Table 1) based on confirmed vs assumed maternity/paternity status, the number of de novo probands, and the phenotypic consistency | (1) To be scored at phenotype highly specific for gene, the patient must meet the PP4 criteria; (2) proband must harbor an additional pathogenic or likely pathogenic variant with the de novo variant | |||
| PP1 | Cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease | Disease specific, strength | N/A | N/A | Segregation in proband plus ≥3 affected relatives | Segregation in proband plus 2 affected relatives | Segregation in proband plus 1 affected relative | Only individuals well documented as having both GT and the variant (phenotype positive, genotype positive) are included when counting segregations; the individuals must be homozygous or compound heterozygous with variants confirmed in trans |
| PP2 | Missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease | N/A | Rule does not apply because benign missense variants are not rare, and the missense constraint z scores for ITGA2B and ITGB3 are <3.09 | |||||
| PP3 | Multiple lines of computational evidence support a deleterious effect on the gene or gene product | Gene specific | N/A | N/A | N/A | N/A | REVEL score of ≥0.7 | |
| PP4 | Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology | Disease specific, strength | N/A | N/A | Meeting criteria (1) and (2) as described for PP4_moderate as well as (3a) absent or reduced (<25% compared with normal) surface expression of αIIbβ3 or (3b) functional flow cytometry to demonstrate lack of fibrinogen-mediated platelet binding to activated αIIbβ3 and (4) full sequencing (all exons and intron-exon boundaries) of both ITGA2B and ITGB3 | (1) At least 1 mucocutaneous bleeding phenotype (eg, epistaxis, petechiae, easy bruising, oral bleeding, gastrointestinal bleeding, or menorrhagia) and (2) normal ristocetin-induced platelet agglutination but minimal to no platelet aggregation with all tested physiological agonists, at a minimum of 2 agonists tested | N/A | Surface expression of αIIbβ3 must be established by either flow cytometry or western blotting |
| PP5 | Reputable source recently reports variant as pathogenic | N/A | Do not use this rule as per SVI recommendations | |||||
| BA1 | Allele frequency is greater than expected for disorder | Disease specific | MAF ≥0.0024 in a gnomAD population cohort | N/A | N/A | N/A | N/A | Cohort must be a continental population with a minimum number of 2000 alleles and the variant present in ≥5 alleles |
| BS1 | Allele frequency is greater than expected for the disorder | Disease specific | N/A | N/A | MAF of 0.00158-0.0024 in a gnomAD population cohort | N/A | N/A | Cohort must have a minimum number of 2000 alleles and the variant present in ≥5 alleles |
| BS2 | Observed in a healthy adult individual for a recessive (homozygous), dominant (heterozygous), or X-linked (hemizygous) disorder, with full penetrance expected at an early age | Disease specific | N/A | N/A | Variant observed in >1 unaffected homozygote | N/A | N/A | Unaffected individuals must have been assessed for GT by at least platelet aggregometry |
| BS3 | Well-established in vitro or in vivo functional studies show no damaging effect on protein function or splicing | Gene specific | N/A | N/A | (1) Normal aggregometry in a transgenic mouse model or (2) both normal surface expression (>75%) and normal fibrinogen binding in either a heterologous cell line or animal model | N/A | N/A | Expression must be measured by flow cytometry or western blot |
| BS4 | Lack of segregation in affected members of a family | Disease specific | N/A | N/A | Lack of segregation in ≥1 affected family member who is well documented as having GT | N/A | N/A | This code should only be applied for phenotype-positive (meeting PP4), genotype-negative family members |
| BP1 | Missense variant in a gene for which primarily truncating variants are known to cause disease | N/A | Rule does not apply because truncating variants do not predominate, and missense variants are a known cause of disease | |||||
| BP2 | Observed in trans with a pathogenic variant for a fully penetrant dominant gene/disorder or observed in cis with a pathogenic variant | None | N/A | N/A | N/A | N/A | Use per original guidelines | This rule can be applied when the variant of interest is observed in cis with a pathogenic variant |
| BP3 | Inframe deletions/insertions in a repetitive region without a known function | None | N/A | N/A | N/A | N/A | Use per original guidelines | ITGB3 has 3 repetitive microsatellite regions, and there are no known repetitive regions in the ITGA2B gene |
| BP4 | Multiple lines of computational evidence suggest no impact on gene/gene product | Gene specific | N/A | N/A | N/A | N/A | REVEL score ≤0.25 | |
| BP5 | Variant found in a case with an alternative molecular basis for disease | N/A | Rule does not apply because an individual with an alternative basis for disease can still be a carrier of an unrelated pathogenic variant in ITGA2B/ITGB3 | |||||
| BP6 | Reputable source recently reports variant as benign | N/A | Do not use this rule as per SVI recommendations | |||||
| BP7 | A synonymous (silent) variant for which splicing prediction algorithms predict no impact to the splice consensus sequence or the creation of a new splice site AND the nucleotide is not highly conserved | None | N/A | N/A | N/A | N/A | Use per original guidelines | |
| ACMG/AMP criteria | Original ACMG/AMP criteria summary | Specification | Standalone | Very strong | Strong | Moderate | Supporting | Comments |
|---|---|---|---|---|---|---|---|---|
| PVS1 | Null variant in a gene where LOF is a known mechanism of disease | Gene specific | N/A | Per modified PVS1 decision tree (Figure 2) with specified regions critical to protein function | N/A | Three critical regions: (1) the cytoplasmic domain of the β3 subunit, (2) the transmembrane domains of αIIbβ3, and (3) the extracellular domains of αIIbβ3 involved in ligand binding | ||
| PS1 | Same amino acid change as a previously established pathogenic variant regardless of nucleotide change | None | N/A | N/A | Use per original guidelines | N/A | N/A | Both variants must be classified using ITGA2B/ITGB3-specified guidelines |
| PS2 | De novo in a patient (maternity and paternity confirmed) with the disease and no family history | Disease specific, strength | N/A | Use the SVI-recommended point system (supplemental Table 1) based on confirmed vs assumed maternity/paternity status, the number of de novo probands, and the phenotypic consistency | (1) To be scored at phenotype highly specific for gene, the patient must meet the PP4 criteria; (2) proband must harbor an additional pathogenic or likely pathogenic variant with the de novo variant | |||
| PS3 | Well-established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product | Gene specific, strength | N/A | N/A | <5% surface expression of αIIbβ3 (measured by flow cytometry or western blot) and/or significantly reduced function (ie, minimal binding to fibrinogen or ligand mimetic antibodies) | 5%-25% surface expression of αIIbβ3 (measured by flow cytometry or western blot) | N/A | All assays must be performed in a heterologous cell line or animal model with expression of the variant of interest from 1 gene (eg, ITGA2B) and the wild type of the opposite gene (eg, ITGB3) |
| PS4 | The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls | N/A | Rule does not apply because of the rarity of disorder and lack of appropriate studies | |||||
| PM1 | Located in a mutational hotspot and/or critical and well-established functional domain without benign variation | N/A | Rule does not apply because of highly polymorphic nature of genes and no known hotspots | |||||
| PM2 | Absent in population databases (or at extremely low frequency if recessive) | Disease specific | N/A | N/A | N/A | N/A | <1 in 10 000 (0.0001) alleles in all gnomAD continental population cohorts; not present in homozygous state | |
| PM3 | For recessive disorders, detected in trans with a pathogenic variant | Disease-specific, Strength | N/A | Use SVI point recommendations (supplemental Table 2); strength may be adjusted based upon confirmation of variant phasing and classification of the second variant | Both variants must be below the PM2 threshold, and both must be classified using ITGA2B/ITGB3-specified guidelines | |||
| PM4 | Protein length changes as a result of inframe deletions/insertions in a nonrepeat region or stop-loss variants | None | N/A | N/A | N/A | Use per original guidelines | N/A | |
| PM5 | Novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before | Strength | N/A | N/A | N/A | Use per original guidelines | Novel missense change at an amino acid residue where a different missense change determined to be likely pathogenic has been seen before | Both variants must be classified using ITGA2B/ITGB3-specified guidelines |
| PM6 | De novo in a patient (maternity and paternity not confirmed) with the disease and no family history | Disease specific, strength | N/A | Use the SVI-recommended point system (supplemental Table 1) based on confirmed vs assumed maternity/paternity status, the number of de novo probands, and the phenotypic consistency | (1) To be scored at phenotype highly specific for gene, the patient must meet the PP4 criteria; (2) proband must harbor an additional pathogenic or likely pathogenic variant with the de novo variant | |||
| PP1 | Cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease | Disease specific, strength | N/A | N/A | Segregation in proband plus ≥3 affected relatives | Segregation in proband plus 2 affected relatives | Segregation in proband plus 1 affected relative | Only individuals well documented as having both GT and the variant (phenotype positive, genotype positive) are included when counting segregations; the individuals must be homozygous or compound heterozygous with variants confirmed in trans |
| PP2 | Missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease | N/A | Rule does not apply because benign missense variants are not rare, and the missense constraint z scores for ITGA2B and ITGB3 are <3.09 | |||||
| PP3 | Multiple lines of computational evidence support a deleterious effect on the gene or gene product | Gene specific | N/A | N/A | N/A | N/A | REVEL score of ≥0.7 | |
| PP4 | Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology | Disease specific, strength | N/A | N/A | Meeting criteria (1) and (2) as described for PP4_moderate as well as (3a) absent or reduced (<25% compared with normal) surface expression of αIIbβ3 or (3b) functional flow cytometry to demonstrate lack of fibrinogen-mediated platelet binding to activated αIIbβ3 and (4) full sequencing (all exons and intron-exon boundaries) of both ITGA2B and ITGB3 | (1) At least 1 mucocutaneous bleeding phenotype (eg, epistaxis, petechiae, easy bruising, oral bleeding, gastrointestinal bleeding, or menorrhagia) and (2) normal ristocetin-induced platelet agglutination but minimal to no platelet aggregation with all tested physiological agonists, at a minimum of 2 agonists tested | N/A | Surface expression of αIIbβ3 must be established by either flow cytometry or western blotting |
| PP5 | Reputable source recently reports variant as pathogenic | N/A | Do not use this rule as per SVI recommendations | |||||
| BA1 | Allele frequency is greater than expected for disorder | Disease specific | MAF ≥0.0024 in a gnomAD population cohort | N/A | N/A | N/A | N/A | Cohort must be a continental population with a minimum number of 2000 alleles and the variant present in ≥5 alleles |
| BS1 | Allele frequency is greater than expected for the disorder | Disease specific | N/A | N/A | MAF of 0.00158-0.0024 in a gnomAD population cohort | N/A | N/A | Cohort must have a minimum number of 2000 alleles and the variant present in ≥5 alleles |
| BS2 | Observed in a healthy adult individual for a recessive (homozygous), dominant (heterozygous), or X-linked (hemizygous) disorder, with full penetrance expected at an early age | Disease specific | N/A | N/A | Variant observed in >1 unaffected homozygote | N/A | N/A | Unaffected individuals must have been assessed for GT by at least platelet aggregometry |
| BS3 | Well-established in vitro or in vivo functional studies show no damaging effect on protein function or splicing | Gene specific | N/A | N/A | (1) Normal aggregometry in a transgenic mouse model or (2) both normal surface expression (>75%) and normal fibrinogen binding in either a heterologous cell line or animal model | N/A | N/A | Expression must be measured by flow cytometry or western blot |
| BS4 | Lack of segregation in affected members of a family | Disease specific | N/A | N/A | Lack of segregation in ≥1 affected family member who is well documented as having GT | N/A | N/A | This code should only be applied for phenotype-positive (meeting PP4), genotype-negative family members |
| BP1 | Missense variant in a gene for which primarily truncating variants are known to cause disease | N/A | Rule does not apply because truncating variants do not predominate, and missense variants are a known cause of disease | |||||
| BP2 | Observed in trans with a pathogenic variant for a fully penetrant dominant gene/disorder or observed in cis with a pathogenic variant | None | N/A | N/A | N/A | N/A | Use per original guidelines | This rule can be applied when the variant of interest is observed in cis with a pathogenic variant |
| BP3 | Inframe deletions/insertions in a repetitive region without a known function | None | N/A | N/A | N/A | N/A | Use per original guidelines | ITGB3 has 3 repetitive microsatellite regions, and there are no known repetitive regions in the ITGA2B gene |
| BP4 | Multiple lines of computational evidence suggest no impact on gene/gene product | Gene specific | N/A | N/A | N/A | N/A | REVEL score ≤0.25 | |
| BP5 | Variant found in a case with an alternative molecular basis for disease | N/A | Rule does not apply because an individual with an alternative basis for disease can still be a carrier of an unrelated pathogenic variant in ITGA2B/ITGB3 | |||||
| BP6 | Reputable source recently reports variant as benign | N/A | Do not use this rule as per SVI recommendations | |||||
| BP7 | A synonymous (silent) variant for which splicing prediction algorithms predict no impact to the splice consensus sequence or the creation of a new splice site AND the nucleotide is not highly conserved | None | N/A | N/A | N/A | N/A | Use per original guidelines | |
MAF, minor allele frequency; N/A, not applicable.
Population data
BA1/BS1: allele frequency greater than expected for disorder.
GT is a rare disease, defined in the United States as affecting <200 000 individuals.19 The exact incidence is unresolved but is estimated at 1 in 1 million worldwide.20 The reported incidence is higher in certain ethnic groups in which consanguinity is common, such as the French Manouche population,21 South Indian Hindus,22 Iranians,23 Iraqi Jews,24 and Jordanian nomadic tribes.25 To set a sufficiently strict requirement for the BA1 criterion, we considered the highest reported prevalence of 1 in 200 000 in the Iranian population.16 The Whiffen/Ware allele frequency threshold calculator26,27 was used with the following parameters: prevalence of 1 in 200 000, complete penetrance, conservative allelic and genetic heterogeneity values of 100%, and 99% confidence. Given these parameters, we recommend application of BA1 to a variant for which ≥1 of the continental population groups in gnomAD28 has an allele frequency ≥0.0024 (0.24%). Accounting for the 2 known genes associated with GT by adjusting genetic heterogeneity to 50%, the BS1 criterion can be applied for a variant with an allele frequency ≥0.0014 (0.14%). The PD-EP also adopted the recommendation by the SVI that the variant be present in at least 5 alleles with a minimum number of 2000 alleles analyzed within the population group to minimize the risk of sequencing error or chance inclusion of an affected individual.
PM2: absent from population databases.
With the availability of ever-larger databases of exome and genome sequence data, the expectation that pathogenic variants will be entirely absent from these databases is not consistent with all diseases. To use the PM2 criterion for ITGA2B/ITGB3, the PD-EP specifies that the variant must be present in <1 in 10 000 alleles in all gnomAD continental population cohorts (allele frequency <0.0001 or 0.01%), and the variant cannot be observed in the homozygous state in any population. This threshold was proposed based on work from Buitrago et al,29 showing that none of the established GT pathogenic variants were identified in the 32 000 alleles studied and suggesting that pathogenic variants have allele frequencies <0.01% in the studied populations. Furthermore, this value is consistent with the highest allele frequency (0.01% in the East Asian population) of the most common known pathogenic GT variant (ITGA2B c.2915dup) in the gnomAD (version 2.1.1) database. The PD-EP also adopted the recommendation by the SVI that the strength of PM2 be modified to PM2_supporting.
Computational and predictive data
PVS1: predicted null variant in a gene where LOF is a known mechanism of disease.
The quantitative or qualitative abnormalities of integrin αIIbβ3, causative of GT, relate to a loss-of-function (LOF) disease mechanism associated with ITGA2B/ITGB3. Variants can cause production of subunits αIIb or β3 to be blocked or interfere with complex formation and/or trafficking.30-33 Although there is an extensive repertoire of missense variants in GT, presumed LOF variants, including nonsense variants, small out-of-frame deletions and insertions, and splice variants that disrupt the reading frame, are also observed. Of the 63 ITGA2B/ITGB3 variants classified in ClinVar as pathogenic or likely pathogenic for GT (accession date 13 February 2020), 26 (41.3%) are presumed LOF, spanning across multiple exons of both genes. For the predicted LOF variants in ITGA2B/ITGB3, the PD-EP recommends the use of the PVS1 decision tree generated by the SVI to guide curators on the applicable PVS1 strength level depending on variant type and features.34 In general, PVS1 can be considered for variants throughout the ITGA2B/ITGB3 genes, because all exons are present in their respective biologically relevant transcript, and no exons are enriched for high-frequency LOF variants in the general population. The PD-EP has modified the decision tree to reflect the particulars of ITGA2B/ITGB3 (Figure 2). The logical flow diagram should be used to evaluate such variants and the text provided here considered to provide context to that diagram.

PVS1 decision tree for ITGA2B/ITGB3 variants. Application of different levels of strength for PVS1 depending on the following factors: type of variant (light blue), prediction of nonsense-mediated decay (NMD; gray), location within a known critical protein domain (green), and size of deletion (yellow). N/A, not applicable.
PVS1 decision tree for ITGA2B/ITGB3 variants. Application of different levels of strength for PVS1 depending on the following factors: type of variant (light blue), prediction of nonsense-mediated decay (NMD; gray), location within a known critical protein domain (green), and size of deletion (yellow). N/A, not applicable.
For any variant that generates a premature stop codon (ie, nonsense, frameshift, or single/multiexon deletion/duplication/skipping), the potential for NMD was considered to follow the general expectation that any premature stop codon occurring before the 3′-most 50 nucleotides of the penultimate exon will lead to NMD.35,36 When NMD is not predicted, one must assess whether the truncated or altered protein product disrupts a region critical to protein function. The PD-EP has identified 3 relevant domains, indicated by experimental evidence to serve a critical role in the shift of αIIbβ3 from a low- to high-affinity state: (1) the cytoplasmic domain of β3, (2) the transmembrane domains of αIIbβ3, and (3) the extracellular domains of αIIbβ3 involved in ligand binding. Binding of intracellular proteins to the cytoplasmic domains of αIIbβ3 prompts an unclasping of the intracellular and transmembrane domains, leading to a conformational change in the extracellular domain (as reviewed by Huang et al37 ). Diverse platelet agonists can alter the β3 cytoplasmic domain and initiate a conformational change,38 which is required for activation. In contrast, loss of the αIIb cytoplasmic domain is not associated with LOF but instead with gain of function.39 As such, only the β3 cytoplasmic domain is considered the first critical region for the interpretation of LOF variants associated with GT. Subsequent separation of the transmembrane domains, the second critical region of αIIbβ3, is also indispensable for ligand-induced signaling.39,40 The third critical region, the extracellular domains involved in ligand binding, includes the β propeller domain of αIIb and the βI domain of β3. Upon activation, the conformation of these 2 subunits is altered, which allows for high-affinity interaction with fibrinogen, among other ligands (as reviewed by Plow et al41 ). Two distinct regions within the βI domain clearly contribute to the ligand binding function,42-45 whereas in the β propeller domain, a role for the 7 N-terminal repeats has been identified.46,47
PM5: novel missense change at an amino acid residue where a different pathogenic missense change has been seen before.
A strength modification was established for PM5: PM5 is used when a different pathogenic missense change has been seen previously at the same residue, and PM5_supporting is used when the previously observed missense change at the same residue is classified as likely pathogenic. Classification of the alternate variant at the same codon must also be based on the ACMG/AMP guidelines as specified by the PD-EP.
PP3: multiple lines of computational evidence support a deleterious effect on the gene or gene product.
This criterion can be used to evaluate either missense variants or those affecting splicing, other than canonical splice sites (PVS1). For missense variants, the PD-EP specifies the use of the Rare Exome Variant Ensemble Learner (REVEL) metapredictor tool; a REVEL score of ≥0.7 is recommended. The REVEL score for an individual missense variant can range from 0 to 1, with a higher score reflecting greater likelihood that the variant is disease causing. Across the genome, REVEL had the best overall performance (P < 10−12) compared with any of 13 individual tool and 7 ensemble methods.48 Substantiation for use of a REVEL score ≥0.7 to apply the PP3 criterion was provided by a set of 50 missense ITGA2B/ITGB3 test variants classified without using in silico predictors, resulting in 14 pathogenic and 12 benign classifications (supplemental Figure 1). Using a REVEL score of ≥0.7, 50% of pathogenic variants met the PP3 criterion (REVEL score, 0.68 ± 0.24), whereas all benign variants (REVEL score ≤0.561) were excluded.
For variants affecting splicing, the PD-EP specified that ≥2 in silico splicing predictors in agreement would qualify for application of the PP3 criterion. This includes missense variants or synonymous variants in the first or last codon of an exon as well as intronic variants other than canonical splice sites (PVS1). For this purpose, the PD-EP used Human Splicing Finder49 (HSF) and Maximum Entropy Scan50 (MaxEntScan) with the standard parameters recommended by each predictor: (1) the threshold for a position to be considered a splice site is ≥65 for HSF and ≥3 for MaxEntScan, (2) a broken splice site is defined as a position that has shifted below the threshold with a difference in score of less than ≤10% for HSF or less than −30% for MaxEntScan, and (3) a new splice site is defined as a position that has shifted above the threshold where the difference in score is >10% for HSF or >30% for MaxEntScan.
BP4: multiple lines of computational evidence suggest no impact on the gene or gene product.
Again, the PD-EP recommends that this criterion can be used to evaluate missense variants with the REVEL metapredictor tool. A REVEL score of ≤0.25 to apply the BP4 criterion was specified based on the same set of 50 missense ITGA2B/ITGB3 test variants (supplemental Figure 1); 75% of benign variants met the BP4 criterion (REVEL score 0.19 ± 0.17), whereas all pathogenic variants (REVEL score ≥0.268) were excluded, and VUSs were present across the range (REVEL score, 0.118-0.972).
Functional data
PS3: well-established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product.
Functional studies can provide strong support of pathogenicity, but only if the experimental assay evaluates the function relevant to the disease mechanism. As such, the PD-EP considered that GT can be caused by either quantitative or qualitative defects in αIIbβ3 that cause loss of ligand (eg, fibrinogen) binding function. All assays must be performed in an animal model or heterologous cell line; analysis of expression and receptor function in patient cells is considered within the PP4 criterion. In those assays with expression in heterologous cell lines, the variant of interest from 1 gene (eg, ITGA2B) is coexpressed with the opposite gene (eg, ITGB3) in its wild-type form.51 There are 2 classes of assays the PD-EP evaluated: those that directly inform on the binding function, and those that report on the absent/reduced expression that leads to impaired ligand binding. Other complementary assays have been reported in the literature but were not considered to fulfill PS3 by the PD-EP. Largely, these assays have been used to evaluate intracellular activities such as subunit synthesis, maturation, interaction, and stability.30,52,53 Although these provide evidence for which step in production is affected, they do not directly inform on the resulting quantitative (protein level) or qualitative (ligand binding) defects on the cell surface.
The effect of the variant on receptor expression or total protein content can be assessed by either flow cytometry or immunoblot,54 and the strength of the PS3 criterion is dependent on the results of the expression assay for the variant compared with a wild-type control. Aside from the variants with qualitative defects, the level of αIIbβ3 surface expression correlates well with the level of ligand binding55 ; thus, a variant with <5% expression is considered to be functionally null because the complex is either absent or amounts are insufficient for meaningful fibrinogen binding to occur. Consequently, an additional functional assay is not required to reach the strong evidence level with expression <5%; however, at 5% to 25% expression, while still consistent with levels observed in type II GT patients, the receptor function may not be insignificant and the variant may or may not also cause a fibrinogen binding defect.56 Therefore, the reduced-strength PS3_moderate criterion should be applied for reduced surface expression (5% to 25%) in the absence of a receptor function assay. A knock-in animal model may be used in place of heterologous cells to meet the PS3 criterion based on the same expression thresholds.
For variants in ITGA2B or ITGB3 where type II (mild quantitative deficiency) or type III (qualitative/functional deficiency) patients are being considered, functional assays in which flow cytometry is used to measure the ability of αIIbβ3 to bind its natural ligand, soluble fibrinogen, or ligand mimetic antibody (eg, PAC-1) are applied.56 However, the use of transfected heterologous cells has limitations in this regard, and as defined by the PD-EP, PS3 evidence for a functionally abnormal receptor requires a positive control with wild-type genes. A variant shown to be functionally abnormal, compared with wild type, by this class of assays meets the strong requirement of PS3. Similarly, and understanding the differences between αIIbβ3 in mouse and humans, a knock-in mouse model may also meet the PS3 criterion, with demonstration that knock-in mouse platelets have either (1) minimal to no αIIbβ3 function, as shown by a fibrinogen (or ligand mimetic antibody, JON/A) binding assay, or (2) abnormal aggregation of mouse platelets similar to that observed in GT patients (PP4), with a normal response to botrocetin.
BS3: well-established in vitro or in vivo functional studies show no damaging effect on protein function or splicing.
Although the quantitative and qualitative aspects of ITGA2B/ITGB3 defects were considered individually for evidence of pathogenicity, both must be considered in combination to determine if functional evidence shows no damaging effect on protein function. A variant expressed in a heterologous cell line may have normal expression, but this alone should not be used as benign evidence, because the variant may still cause a qualitative defect. Accordingly, the PD-EP specifies that the BS3 criterion can be satisfied by either (1) a heterologous cell line that exhibits both normal expression and normal binding (description of these assays provided in PS3) or (2) a knock-in mouse model with normal platelet aggregometry or demonstration of both normal expression and normal ligand binding.
Segregation data
PP1: cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease.
Cosegregation of the variant of interest with disease in 1 proband plus 1 affected relative is considered sufficient to meet the PP1 criterion at the supporting level. Of note, only individuals well documented as having both GT and the variant are included when counting segregations. All GT individuals within a family must be of the same genotype, either homozygous or compound heterozygous (with variants confirmed in trans). Additionally, the PD-EP has adopted the approach taken by various ClinGen EPs6,57-60 and supported by both the SVI and the Clinical Sequencing Exploratory Research Consortium61 that additional meioses support higher levels of evidence. Therefore, the PD-EP specifies that a proband plus 2 affected relatives is sufficient for PP1_moderate, and a proband plus ≥3 affected relatives meets PP1_strong.
BS4: lack of segregation in affected members of a family.
Lack of segregation of a variant with a phenotype can provide strong benign evidence but should be used prudently. For family members who have a bleeding phenotype but are negative for the variant in question, there is the possibility of an alternative explanation of the mucocutaneous bleeding. For instance, rare coinheritance of GT with other bleeding disorders has been reported.62 To ensure only families with true lack of segregation (≥2 affected family members with at least 1 affected individual not carrying the variant of interest) are included, the PD-EP specifies that all affected family members must be well documented as having GT based on both bleeding phenotype and appropriate laboratory values (meeting the PP4_moderate criterion).
De novo data
PS2/PM6: de novo occurrence.
In GT patients, as with other disorders inherited in an autosomal recessive manner, de novo variants are extremely rare; however, de novo occurrences have been reported.63 In consideration of these rare cases, the PM6 de novo, maternity and paternity not confirmed, and PS2 de novo, paternity and maternity confirmed, criteria were specified for GT with adoption of SVI recommendations (supplemental Table 1). Using the SVI-recommended approach, the level of strength for PM6/PS2 is based on confirmed vs assumed maternity/paternity status, the number of de novo probands, and the phenotypic consistency. To be scored at the highest level of phenotypic consistency, phenotype highly specific for gene, the PD-EP has specified that the patient must meet the PP4_moderate criteria. Based on ACMG/AMP guidance, family history must be consistent with a de novo event (ie, both parents must have tested negative for the variant), and further PD-EP specification requires that the PS2/PM6 criteria are only applied when the proband harbors a second pathogenic or likely pathogenic variant (as classified by the PD-EP–specified ACMG/AMP guidelines) in addition to the de novo variant.
Allelic data
PM3: for recessive disorders detected in trans with a pathogenic variant in an affected patient.
When 2 heterozygous variants are identified in either ITGA2B or ITGB3, and 1 variant is known to be pathogenic, then determining that the other variant in the same gene is in trans, rather than in cis, can be considered evidence for pathogenicity of the latter variant. Again, a point-based system was recommended by the SVI in which each proband with the variant of interest is awarded a point value, and the combined point value determines the appropriate strength level (supplemental Table 2). The strength of the in trans observation is based upon confirmation of variant phasing and classification of the variant occurring on the other allele (based on the PD-EP–specified ACMG/AMP guidelines). Additionally, application of PM3 is contingent on both variants (or the homozygous variant) in the proband occurring at sufficiently rare allele frequencies that meet the PM2 threshold (<0.0001 or <0.01%).
Phenotypic data
PP4: patient’s phenotype or family history is highly specific for a disease with a single genetic etiology.
The pathogenic criterion PP4 can be applied when the patient’s phenotype meets both clinical and laboratory criteria for GT. In addition to a diagnosis of GT established by a clinician, the PD-EP has defined 3 criteria for assessing the relevance of the PP4 criterion: (1) at least 1 mucocutaneous bleeding phenotype (eg, epistaxis, petechiae, easy bruising, oral bleeding, gastrointestinal bleeding, or menorrhagia); (2) normal ristocetin-induced platelet agglutination but minimal to no platelet aggregation with all tested physiological agonists, at a minimum of 2; and (3) demonstration of a quantitative or qualitative defect of αIIbβ3 in patient cells by either absent or reduced (≤25% compared with normal) surface expression of αIIbβ3 established by flow cytometry or total protein measured by immunoblotting or functional flow cytometry to demonstrate lack of fibrinogen binding to activated αIIbβ3. Fulfillment of all 3 criteria provides sufficient clinical and laboratory detail to satisfy PP4_strong (Figure 3). An additional requirement for the use of PP4_strong is full coding sequencing (all exons and intron-exon boundaries) of both ITGA2B and ITGB3 to minimize the possibility that other plausibly causative variants are present. Given the high specificity of the combined clinical and laboratory phenotype, in combination with the high penetrance of GT, a Bayesian analysis was completed to demonstrate that this is a strong level of evidence. The PD-EP determined that the likelihood ratio that biallelic variants identified in a phenotype-positive individual are pathogenic is 91:1 (95% CI, 32:1-255:1), consistent with the >18.7:1 ratio required for a strong level of evidence in the Bayesian model of the classification framework (supplemental Figure 2).18 For individuals lacking criterion 3, demonstration of a quantitative or qualitative defect in αIIbβ3 or with only partial sequencing, PP4 would be downgraded to a moderate level of evidence. Accompanying normal results are expected for routine tests of a patient with abnormal bleeding. Evaluation of the peripheral blood smear by light microscopy should show normal platelet count and platelet size, and coagulation screening tests such as prothrombin and activated thromboplastin times should be normal. Although these normal results are expected for a GT patient, the PD-EP does not require this information to apply PP4 in variant interpretation.
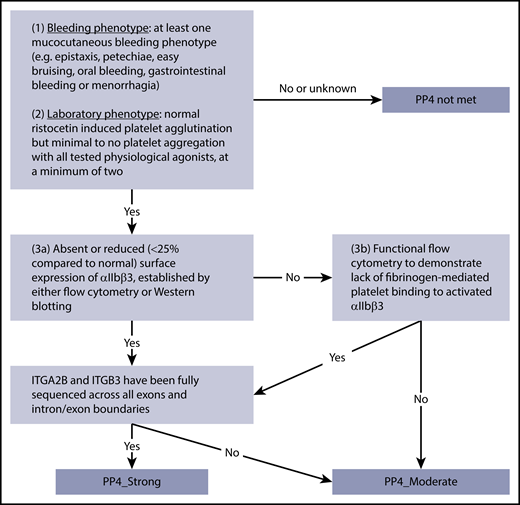
PP4 decision tree for GT patients. Application of different levels of strength for PP4 depending on bleeding and laboratory phenotypes, analysis of αIIbβ3 expression or function, and sequencing coverage.
PP4 decision tree for GT patients. Application of different levels of strength for PP4 depending on bleeding and laboratory phenotypes, analysis of αIIbβ3 expression or function, and sequencing coverage.
BS2: observed in a healthy adult individual with full penetrance expected at an early age.
Observation of the homozygous state of a particular variant in a healthy adult individual is considered strong evidence for a benign interpretation, but the healthy status must be carefully considered. Children are often diagnosed with GT before the age of 5 years, but because the diagnosis is rare and generally undertaken only in specialist centers, it may go undiagnosed or misdiagnosed.64 As such, the PD-EP has specified this criterion such that the variant must be identified in >1 homozygous unaffected individual who has been assessed for GT by at least platelet aggregometry; population data (ie, gnomAD) are not sufficient as evidence. This criterion will aid in assessment of variants identified in the setting of gene panel testing, where an individual with a different bleeding or platelet phenotype is found to be homozygous for a variant in either ITGA2B or ITGB3.
Criteria considered not applicable
Of the original 28 published ACMG/AMP criteria, 4 of the 16 criteria for pathogenic evidence and 3 of the 12 criteria for benign evidence were deemed not applicable for ITGA2B/ITGB3 with respect to GT. Because of the rarity of GT and the lack of appropriate studies to validate the criterion, use of PS4 (prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls) is not recommended. With regard to both PM1 (located in mutational hotspot and/or critical domain without benign variation) and PP2 (missense variant in a gene that has a low rate of benign missense variation), ITGA2B and ITGB3 are highly polymorphic29 with benign variants present across all domains.65,66 Truncating variants make up only a small portion of GT disease-causing variants, whereas missense variants make up the largest class at 47.6% of all pathogenic or likely pathogenic ITGA2B/ITGB3 variants in ClinVar; as such, BP1 (missense variant in a gene with primarily truncating variants) does not apply. The PD-EP also determined that the BP5 criterion (variant found in a case with an alternate molecular basis for disease) should not be used for GT because it is a recessive disorder, and an individual with an alternative basis for a given clinical phenotype can still be a carrier of a single unrelated pathogenic variant in ITGA2B/ITGB3. Furthermore, having GT as the result of biallelic pathogenic ITGA2B variants would not preclude an individual from being a carrier of a single pathogenic ITGB3 variant. In the case of PP5 (reputable source reports as pathogenic) and BP6 (reputable source reports as benign), these criteria were removed based on recommendation from the SVI.67
Validation of the GT ITGA2B/ITGB3-specific ACMG/AMP variant interpretation guidelines
To ensure the clarity and applicability of rule specifications, the PD-EP evaluated variants encompassing a wide breadth of evidence. The most used criteria for variant interpretation were PM2, PP4, PM3, and PP3 (Figure 4). In addition, 16 variants were predicted to be LOF, meeting the PVS1 criterion, and 15 variants have data from in vitro functional studies, allowing the use of PS3. The commonly used benign criteria were allele frequency criterion BA1 or BS1 and computational/predictive criterion BP7 or BP4. Additionally, 4 variants had in vitro functional studies meeting BS3, and 2 variants were identified in cis (BP2) with pathogenic variants.
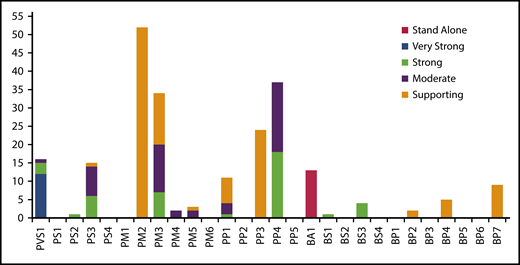
Summary of the variant interpretation criteria applied to the 70 ITAG2B/ITGB3 validation variants. The frequency with which PD-EP–specified ACMG/AMP criteria were applied during the variant pilot. Several criteria were applied at varying levels of strength, as indicated by shading (legend on the right). A majority of rules were applied at least once; however, no relevant variants were identified for application of PS1, PM6, BS2, BS4, or BP3 criteria.
Summary of the variant interpretation criteria applied to the 70 ITAG2B/ITGB3 validation variants. The frequency with which PD-EP–specified ACMG/AMP criteria were applied during the variant pilot. Several criteria were applied at varying levels of strength, as indicated by shading (legend on the right). A majority of rules were applied at least once; however, no relevant variants were identified for application of PS1, PM6, BS2, BS4, or BP3 criteria.
For pilot testing, 14 benign or likely benign variants, 17 VUSs, 33 pathogenic or likely pathogenic variants, and 6 conflicting variants (within ClinVar or between ClinVar and initial expert assertions) were recommended for inclusion by PD-EP members based on initial expert-based assertions of pathogenicity before the use of GT-specified guidelines (supplemental Tables 3 and 4). Using the ITGA2B/ITGB3-specific guidelines, 15 validation variants were classified by the PD-EP as benign or likely benign, 14 were VUSs, and 41 were pathogenic or likely pathogenic (Figure 1; supplemental Tables 3 and 4). A list of all pilot variants, the variant classification of the ClinVar submitters, and the initial and final classifications made by the PD-EP (with the criteria used) are presented in supplemental Tables 3 and 4. Initial expert assertions (made before disease/gene specification of the ACMG/AMP guidelines) for all 70 pilot variants were compared with the final classifications based on GT-specific guidelines. There was 71% clinical concordance between initial expert assertions and classification based on the PD-EP–specified criteria (Figure 5). Thirty of 34 variants initially asserted as pathogenic or likely pathogenic by the PD-EP submitters remained at that assertion after applying the GT-specific guidelines. The remaining 4 variants were downgraded to VUSs because of insufficient pathogenic evidence. Thirteen of 16 variants initially asserted as benign or likely benign remained at that assertion after applying the GT-specific guidelines. The remaining 3 variants were reclassified as VUSs because of insufficient and conflicting evidence. Twenty variants were initially interpreted as VUSs by our EP members; 2 of these 20 were classified as benign or likely benign based on population frequencies, 7 remained VUSs because of insufficient evidence, and 11 were classified as pathogenic or likely pathogenic. Applying the GT-specific ACMG/AMP criteria to the pilot variants resulted in a reduction in VUS classification from 29% to 20%.

Comparison of initial expert assertions and final PD-EP classifications of the 70 ITAG2B/ITGB3 validation variants. The total height of each bar represents the number of variants with each initial assertion. The colored segments of each bar represent the final PD-EP classification (legend on the right).
Comparison of initial expert assertions and final PD-EP classifications of the 70 ITAG2B/ITGB3 validation variants. The total height of each bar represents the number of variants with each initial assertion. The colored segments of each bar represent the final PD-EP classification (legend on the right).
In addition to validating the PD-EP ITGA2B/ITGB3 GT guideline specifications against initial expert assertions, ClinVar interpretations were also considered. Of the 70 ITGA2B/ITGB3 pilot variants, 34 had interpretations in ClinVar, 76% of which were in clinical concordance with the classification made with PD-EP–specified guidelines (supplemental Tables 3 and 4), increasing to 100% concordance for variants with nonconflicting interpretations from multiple submitters in ClinVar. Nine pilot variants had clinically discordant interpretations either within ClinVar or between ClinVar and the final PD-EP classifications. One pilot variant with ClinVar-conflicting interpretations (likely benign and VUS) was classified by the PD-EP as benign using the GT-specific guidelines, and 4 ClinVar VUSs were reclassified to pathogenic or likely pathogenic and 2 to benign or likely benign (Table 2). The 2 remaining discordant variants, with likely pathogenic and likely benign ClinVar interpretations, respectively, were not able to reach classification with the PD-EP–specified guidelines and were considered VUSs. Overall, of the 34 variants with ClinVar interpretations, 1 with conflicting interpretations and 6 with VUS interpretations were reclassified to the appropriate pathogenic, likely pathogenic, benign, or likely benign category (21%), whereas only 2 variants were moved to the VUS classification (6%).
ITGA2B/ITGB3 variants with discordant initial and final classifications
| Gene | cDNA | Amino acid | ClinVar assertion | Initial expert assertion | Final PD-EP classification | Criteria applied |
|---|---|---|---|---|---|---|
| ClinVar/initial assertion conflicting | ||||||
| ITGA2B | 3060+2T>C | VUS | Pathogenic | Likely pathogenic | PVS1_S, PM2_P, PM3_P, PP4_M | |
| ITGA2B | 2965G>A | Ala989Thr | VUS | Likely benign | Likely benign | BS1, BP2, BP4 |
| ITGA2B | 891+12del | VUS | Benign | Benign | BA1, BP7 | |
| ITGB3 | 565C>T | Pro189Ser | Likely pathogenic | VUS | Pathogenic | PS3_M, PM2_P, PM3_S, PP1, PP3, PP4 |
| ITGB3 | 1960G>A | Glu654Lys | VUS and likely benign | VUS | Benign | BA1 |
| ITGB3 | 1902C>T | Cys634= | Likely benign | VUS | Benign | BA1, BP7 |
| Reclassified from VUS | ||||||
| ITGA2B | 1234G>A | Gly412Arg | VUS | VUS | Pathogenic | PM2_P, PM3_S, PP3, PP4_S |
| ITGA2B | 257T>C | Leu86Pro | N/A | VUS | Pathogenic | PS3_M, PM2, PM3_P, PP3, PP4_S |
| ITGA2B | 460_462del | Glu154del | N/A | VUS | Likely pathogenic | PM2_P, PM3_P, PM4, PP4_M |
| ITGB3 | 362-1G>A | VUS | VUS | Likely pathogenic | PVS1, PM2_P | |
| ITGB3 | 187C>T | Arg63Cys | VUS | VUS | Likely pathogenic | PS3_M, PM2_P, PM3_P, PP3, PP4_M |
| ITGB3 | 448A>G | Met150Val | N/A | VUS | Likely pathogenic | PM2_P, PM3, PP3, PP4_M |
| ITGB3 | 629G>C | Cys210Ser | N/A | VUS | Likely pathogenic | PS3, PM2_P, PP1, PP3, PP4_M |
| ITGB3 | 1458C>G | Cys486Trp | N/A | VUS | Likely pathogenic | PM2_P, PM3, PP3, PP4_S |
| ITGB3 | 1594T>C | Cys532Arg | N/A | VUS | Likely pathogenic | PM2_P, PM3, PP3, PP4_S |
| ITGB3 | 1595G>A | Cys532Tyr | N/A | VUS | Likely pathogenic | PM2_P, PM3_P, PM5_P, PP3, PP4_M |
| Reclassified to VUS | ||||||
| ITGA2B | 2852_2853delinsC | Asp951AlafsTer? | N/A | Likely pathogenic | VUS | PVS1_S, PM2_P |
| ITGA2B | 889G>C | Ala297Pro | N/A | Likely pathogenic | VUS | PM2_P, PM3_P, PP1, PP4_M |
| ITGA2B | 1821G>A | Thr607= | Likely benign | Likely benign | VUS | PP3 |
| ITGA2B | 2728-19T>C | N/A | Likely benign | VUS | BP7, PM2_P | |
| ITGB3 | 1366A>C | Thr456Pro | Likely pathogenic | Pathogenic | VUS | PM2_P, PM3_P |
| ITGB3 | 953 T>C | Leu318Ser | N/A | Likely pathogenic | VUS | PM2_P, PM3_P, PP3, PP4_M |
| ITGB3 | 1125+29G>C | N/A | Benign | VUS | BP7, PM2_P |
| Gene | cDNA | Amino acid | ClinVar assertion | Initial expert assertion | Final PD-EP classification | Criteria applied |
|---|---|---|---|---|---|---|
| ClinVar/initial assertion conflicting | ||||||
| ITGA2B | 3060+2T>C | VUS | Pathogenic | Likely pathogenic | PVS1_S, PM2_P, PM3_P, PP4_M | |
| ITGA2B | 2965G>A | Ala989Thr | VUS | Likely benign | Likely benign | BS1, BP2, BP4 |
| ITGA2B | 891+12del | VUS | Benign | Benign | BA1, BP7 | |
| ITGB3 | 565C>T | Pro189Ser | Likely pathogenic | VUS | Pathogenic | PS3_M, PM2_P, PM3_S, PP1, PP3, PP4 |
| ITGB3 | 1960G>A | Glu654Lys | VUS and likely benign | VUS | Benign | BA1 |
| ITGB3 | 1902C>T | Cys634= | Likely benign | VUS | Benign | BA1, BP7 |
| Reclassified from VUS | ||||||
| ITGA2B | 1234G>A | Gly412Arg | VUS | VUS | Pathogenic | PM2_P, PM3_S, PP3, PP4_S |
| ITGA2B | 257T>C | Leu86Pro | N/A | VUS | Pathogenic | PS3_M, PM2, PM3_P, PP3, PP4_S |
| ITGA2B | 460_462del | Glu154del | N/A | VUS | Likely pathogenic | PM2_P, PM3_P, PM4, PP4_M |
| ITGB3 | 362-1G>A | VUS | VUS | Likely pathogenic | PVS1, PM2_P | |
| ITGB3 | 187C>T | Arg63Cys | VUS | VUS | Likely pathogenic | PS3_M, PM2_P, PM3_P, PP3, PP4_M |
| ITGB3 | 448A>G | Met150Val | N/A | VUS | Likely pathogenic | PM2_P, PM3, PP3, PP4_M |
| ITGB3 | 629G>C | Cys210Ser | N/A | VUS | Likely pathogenic | PS3, PM2_P, PP1, PP3, PP4_M |
| ITGB3 | 1458C>G | Cys486Trp | N/A | VUS | Likely pathogenic | PM2_P, PM3, PP3, PP4_S |
| ITGB3 | 1594T>C | Cys532Arg | N/A | VUS | Likely pathogenic | PM2_P, PM3, PP3, PP4_S |
| ITGB3 | 1595G>A | Cys532Tyr | N/A | VUS | Likely pathogenic | PM2_P, PM3_P, PM5_P, PP3, PP4_M |
| Reclassified to VUS | ||||||
| ITGA2B | 2852_2853delinsC | Asp951AlafsTer? | N/A | Likely pathogenic | VUS | PVS1_S, PM2_P |
| ITGA2B | 889G>C | Ala297Pro | N/A | Likely pathogenic | VUS | PM2_P, PM3_P, PP1, PP4_M |
| ITGA2B | 1821G>A | Thr607= | Likely benign | Likely benign | VUS | PP3 |
| ITGA2B | 2728-19T>C | N/A | Likely benign | VUS | BP7, PM2_P | |
| ITGB3 | 1366A>C | Thr456Pro | Likely pathogenic | Pathogenic | VUS | PM2_P, PM3_P |
| ITGB3 | 953 T>C | Leu318Ser | N/A | Likely pathogenic | VUS | PM2_P, PM3_P, PP3, PP4_M |
| ITGB3 | 1125+29G>C | N/A | Benign | VUS | BP7, PM2_P |
Criteria applied with a modified strength are denoted by the criteria followed by _P for supporting, _M for moderate, _S for strong, and _VS for very strong.
cDNA, complementary DNA; N/A, not applicable.
Discussion
Accurate detection and appropriate interpretation of pathogenicity of genetic variants are crucial in the diagnosis, management, and counseling of patients with inherited disorders. The large number of genomic variants in human populations renders the assessment of variant pathogenicity a highly complex and challenging task. Rather than relying on individual laboratories’ internally developed variant interpretation criteria, the 2015 ACMG/AMP publication represented a great leap forward in the field of clinical genetics by providing a set of standardized guidelines. Nevertheless, even armed with these guidelines, significant improvements in the consistency of variant classifications made by different laboratories were not immediately achieved. The inconsistency was revealed in a Clinical Sequencing Exploratory Research Consortium study in 2016,2 which found that inconsistency in variant classification could be at least partially attributed to the degree of subjectivity allowed by the criteria. In the conclusion, recommendations were made to help increase the consistency in the use of ACMG/AMP rules, and most of these involved disease- or gene-specific criteria.2 Subsequently, ClinGen established EPs to adapt ACMG/AMP rules for specific genes or diseases. One of the pioneer EP efforts was made by the ClinGen Cardiovascular Clinical Domain Working Group in adapting rules for MYH7-associated inherited cardiomyopathies. These adapted rules, together with expert review and clinical judgment, successfully increased specificity for interpreting MYH7 variants.6
In the domain of platelet disorders, the ClinGen PD-EP adopted a similar overall framework as the MYH7 project including 3 phases: development of draft modifications, validation through pilot variants, and refinement and finalization of the adapted rules for ITGA2B/ITGB3. The set of rule specifications described herein serve as the expert recommendations for interpreting ITGA2B/ITGB3 variants with respect to GT in the future. The variants curated and classified by the PD-EP will be submitted to ClinVar with a 3-star rating denoted.
During the validation process, we came across several challenges related to the applicability of certain criteria, and these limitations may also be encountered by others performing variant curation. First, the clinical phenotype and testing results of some patients reported in the literature are incomplete, and therefore, the diagnosis of GT could not be clearly ascertained. Although it is possible to contact the authors to request additional information for verification, the PD-EP decided to not pursue this option on a regular basis because it does not mimic the routine practices in a majority of clinical laboratories during variant curation. Therefore, the evidence from these publications was evaluated but often was insufficient to apply certain rules. For this reason, the PD-EP recommends in future workup of patients with suspected GT to acquire the clinical and laboratory information needed for definitively applying the adapted variant classification rules (supplemental Table 5). Second, the PD-EP established relatively stringent acceptance criteria for the types of laboratory or experimental studies to ensure the quality of evidence. Because some older studies in the literature did not meet these criteria, they were deemed ineligible for use during variant curation. The knowledge of laboratory or experimental methods in combination with expert judgment played an important role in the evaluation of this evidence.
Although a majority (71%) of pilot variants had final classifications in clinical concordance with initial expert assertions, several were reclassified (Table 2). Three variants, identified in heterozygosity in patients not diagnosed with GT, were initially asserted as benign or likely benign but were reclassified to VUSs, including 1 intronic variant each in ITGA2B (c.2728-19T>C) and ITGB3 (c.1125+29G>C). Both of these variants may ultimately prove to be benign; however, they are currently classified as VUSs, meeting only the BP7 (no predicted effect on splicing and not a highly conserved nucleotide) and PM2_supporting (rare in population databases) criteria, highlighting the need for benign evidence beyond predictive information to make a clinically significant classification for variants not meeting benign population thresholds (BA1/BS1). For this purpose, among other benign criteria, the PD-EP has specified how functional studies can be used for the application of BS3. Four variants, identified in GT patients and initially asserted as pathogenic or likely pathogenic, were also reclassified to VUSs. The ITGA2B variant c.2852_2853delinsC met the PVS1_strong criterion because of an alteration of the entire transmembrane domain and was absent from population databases (PM2_supporting); however, the variant had insufficient clinical information to fulfill the PP4 of PM3 criteria. The remaining 3 were missense variants in ITGA2B (Ala297Pro) and ITGB3 (Thr456Pro and Leu318Ser) with clinical information provided on the patients; however, they had no reported functional studies meeting the PS3 criteria. This highlights the importance of functional studies in variant classification, particularly for missense variants found in only 1 proband, as commonly occurs in GT.
Conversely, 11 variants initially considered VUSs were reclassified as pathogenic or likely pathogenic. One splice variant, ITGB3 c.362-1G>A, was initially considered a VUS based on the fact that this variant has only been observed in heterozygosity in an ostensibly healthy population; however, PD-EP–specified thresholds for PM2 allowed for application of PVS1 and PM2_supporting, classifying the variant as likely pathogenic. An additional variant was an inframe single amino acid deletion, and the remaining 9 variants were all missense. These variants largely benefited from additional reports in the literature and GT database. There were reports in multiple probands that allowed for PM3 to be applied with an adjustable strength for most variants (9 of 10) and several classifications (4 of 10) were further aided by functional studies meeting the specifications of the PD-EP for the PS3 criterion. The establishment of these standards for variant interpretation should serve to stimulate the field to perform more detailed evaluations of variants and patients and fully report these data so that they can be used in variant interpretation.
In summary, the ClinGen PD-EP effort is the first systematic, rigorous, and validated classification of GT variants. We generated variant interpretation rules that are specific for autosomal recessive GT. Moving forward, these new rules should be taken as the reference for assessment of presumed GT patients, although we acknowledge that technological advances may require us to update the rules in the future. As the next steps, the PD-EP will curate and classify the rest of the ClinVar GT variants, which will be denoted with a 3-star rating and receive the US Food and Drug Administration approval label. The PD-EP also plans to develop rule specifications for the gain-of-function ITGA2B/ITGB3 variants, which cause autosomal dominant macrothrombocytopenia, encompassing a different phenotype, inheritance pattern, and functional defect. After completion of the ITGA2B/ITGB3 variant curation project, the PD-EP plans to adapt the ACMG/AMP rules for additional platelet disorders, including Bernard-Soulier syndrome. In the long term, we hope that these efforts will help hematologists to better interpret clinical genetic panels and therefore improve diagnosis and treatment of platelet disorders.
The ClinGen Expert Panel curated data is deposited in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/submitters/507662/).
Acknowledgments
The ClinGen PD-EP thanks Leslie Biesecker, Steven Harrison, and other members of the ClinGen SVI Working Group for their thoughtful review of these rule specifications and feedback. The EP also thanks Nigel S. Key, chair of the ClinGen Hemostasis/Thrombosis Clinical Domain Working Group, and executive members for their guidance through this process and Meera Jairath for her assistance curating the literature and Dara McDougal for her assistance with working group coordination. Results provided in this publication were generated by ASH in collaboration with the University of North Carolina at Chapel Hill, a National Institutes of Health–funded Clinical Genome Resource grant award recipient.
This work was supported by the National Human Genome Research Institute, National Institutes of Health (grant U41HG009650).
Authorship
Contribution: All authors contributed to the development of the ClinGen PD-EP rule specifications, variant curation review, and manuscript edits; J.E.R. and S.M. were the primary curators for the pilot variant curation study; J.E.R. and B.M.Z. took the lead on manuscript preparation; and the manuscript was edited by all authors.
Conflict-of-interest disclosure: B.R.B., S.N.D., M.L., and J.P.B. work for clinical diagnostic laboratories that offer testing for GT. B.M.Z. is involved in the curation and interpretation of exome sequencing in patients with inherited platelet disorders, as well as the development of an NGS diagnostic gene panel for inherited platelet disorder. M.P.L. is an advisory board member for Octapharma and Shionogi, is a consultant for Amgen, Novartis, Shionogi, Dova, Principia, Argenx, Rigel, and Bayer, and has received research funding from Sysmex, Novartis, Rigel, and AstraZeneca. The remaining authors declare no competing financial interests.
A complete list of the members of the ClinGen Platelet Disorder Variant Curation Expert Panel appears in “Appendix.”
Correspondence: Jorge Di Paola, Department of Pediatrics, Washington University in St. Louis, 660 S Euclid Ave, Campus Box 8208, St. Louis, MO 63110; e-mail: dipaolaj@wustl.edu; and Wolfgang Bergmeier, University of North Carolina at Chapel Hill, 116 Manning Dr, 8212A MEJ Building, Chapel Hill, NC 27599-7035; e-mail: bergmeie@email.unc.edu.
Appendix
The members of the ClinGen Platelet Disorders Variant Curation Expert Panel are: Jorge Di Paola, Wolfgang Bergmeier, Brian R. Branchford, Paul Bray, Samya Chakravorty, Stefanie N. Dugan, Annabelle Frantz, Kathleen Freson, Paula G. Heller, Walter H. A. Kahr, Michele P. Lambert, Kristy Lee, Claire Lentaigne, Minjie Luo, Lori Luchtman-Jones, Shannon McNulty, Andrew Mumford, Madhu Ouseph, Juliana Perez Botero, Jose Rivera, Matt Rondina, Justyne E. Ross, Gabriella Ryan, Sarah Westbury, and Bing M. Zhang.

