

Key Points
Survival after transplantation for NMDs is excellent beyond the first 2 years post-HCT.
Cumulative incidence of SNs is low; however, there is an increased risk in those with FA or marrow failure.

Abstract
We examined the risk of subsequent neoplasms (SNs) and late mortality in children and adolescents undergoing allogeneic hematopoietic cell transplantation (HCT) for nonmalignant diseases (NMDs). We included 6028 patients (median age, 6 years; interquartile range, 1-11; range, <1 to 20) from the Center for International Blood and Marrow Transplant Research (1995-2012) registry. Standardized mortality ratios (SMRs) in 2-year survivors and standardized incidence ratios (SIRs) were calculated to compare mortality and SN rates with expected rates in the general population. Median follow-up of survivors was 7.8 years. Diagnoses included severe aplastic anemia (SAA; 24%), Fanconi anemia (FA; 10%), other marrow failure (6%), hemoglobinopathy (15%), immunodeficiency (23%), and metabolic/leukodystrophy syndrome (22%). Ten-year survival was 93% (95% confidence interval [95% CI], 92% to 94%; SMR, 4.2; 95% CI, 3.7-4.8). Seventy-one patients developed SNs (1.2%). Incidence was highest in FA (5.5%), SAA (1.1%), and other marrow failure syndromes (1.7%); for other NMDs, incidence was <1%. Hematologic (27%), oropharyngeal (25%), and skin cancers (13%) were most common. Leukemia risk was highest in the first 5 years posttransplantation; oropharyngeal, skin, liver, and thyroid tumors primarily occurred after 5 years. Despite a low number of SNs, patients had an 11-fold increased SN risk (SIR, 11; 95% CI, 8.9-13.9) compared with the general population. We report excellent long-term survival and low SN incidence in an international cohort of children undergoing HCT for NMDs. The risk of SN development was highest in patients with FA and marrow failure syndromes, highlighting the need for long-term posttransplantation surveillance in this population.
Introduction
Allogeneic hematopoietic cell transplantation is a curative treatment option for pediatric and adolescent patients with nonmalignant diseases (NMDs). Despite continued advances in the field of HCT and an increasing number of long-term survivors, treatment-related mortality and late effects remain a challenge. A significant cause of morbidity after HCT is the development of subsequent neoplasms (SNs).1 Among children receiving transplants for primary malignancies, exposure to cytotoxic therapy or radiation during initial cancer treatment followed by myeloablative conditioning before HCT increases the likelihood of SN posttransplantation. (HCT). Presumably, children and adolescents receiving transplants for NMDs are at lower risk for SNs than might be expected in children undergoing HCT for malignant diseases. However, although a majority of these patients are not exposed to prior chemotherapy or radiation, graft-versus-host disease (GVHD), immunosuppressive therapy, and genetic predisposition may also increase the risk of SN posttransplantation.
Numerous studies have described SN incidence in children and adolescents undergoing HCT for the treatment of primary malignancies2 ; however, few have examined SN risk in those undergoing transplantation for NMDs.3 Given the increasing use of HCT for children with NMDs, data regarding these long-term outcomes are of high importance. A recent report from the National Institutes of Health Hematopoietic Cell Transplantation Late Effects Initiative highlighted gaps in the current literature regarding SNs after HCT.1 Authors cited a need for longitudinal studies and for comparison of SN risk after HCT with the risk of new malignancies in the general population.4
The objectives of this study were to examine the incidence of posttransplantation SNs and report the risk of death after 2 years in an international cohort of pediatric and adolescent patients with bone marrow failure disorders, hemoglobinopathies, immunodeficiency syndromes, inborn errors of metabolism, and leukodystrophies. We specifically examined whether patients had an increased risk of developing a new malignancy at any time point and an increased risk of late mortality compared with the general population of age- and sex-matched controls. Here we report contemporary data on a diverse patient population to both inform decision making and contribute to consensus guidelines about posttransplantation follow-up surveillance in children and adolescents receiving transplants for NMDs.5
Patients and methods
Data source
The Center for International Blood and Marrow Transplant Research (CIBMTR), a research collaboration between the Medical College of Wisconsin and the National Marrow Donor Program, collects data from >450 transplantation centers worldwide. Participating centers contribute data about individual patients, their exposures, and their outcomes. Transplant essential data are collected on all patients and include information on demographics, primary disease, transplantation characteristics, and posttransplantation outcomes (including SNs, relapse, survival, and cause of death). Observational studies conducted by the CIBMTR are performed in compliance with all applicable federal regulations pertaining to the protection of human research participants.
Study cohort
The study cohorts included in analyses of survival and SNs are presented in Figure 1. Children and adolescents undergoing first HCT for treatment of Fanconi anemia (FA), severe aplastic anemia (SAA), other marrow failure disorders, sickle cell disease (SCD), thalassemia, primary immunodeficiency syndromes (PIDs), histiocytic disorders, or inborn errors of metabolism or leukodystrophy with data reported to the CIBMTR were eligible for inclusion. A total of 6028 patients age <21 years who underwent transplantation between 1995 and 2012 met inclusion criteria (Figure 1).
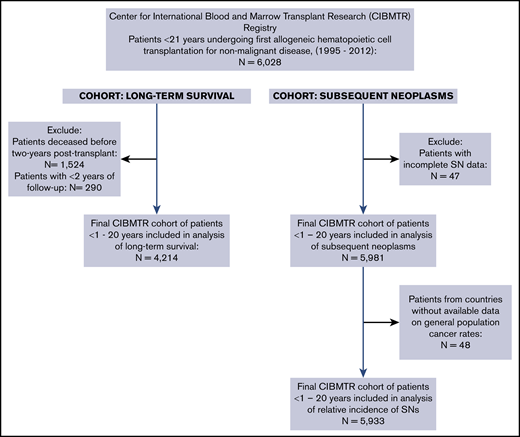
For analysis of late mortality and long-term survival, patients who died (n = 1524; 25%) or who were lost to follow-up before 2 years (n = 251; 5%) were excluded. For analysis of SNs, incidence of SNs for each NMD group was determined from the time of transplantation to the time of event. Patients without information on SNs and those who had an SN diagnosis date that preceded the date of HCT were excluded. An additional 48 patients were excluded from calculation of SN standardized incidence ratios (SIRs) because general population cancer rates were not available for their countries of origin; these included patients from South Africa (n = 33), Venezuela (n = 7), Mexico (n = 7), and Croatia (n = 1). Posttransplantation lymphoproliferative disorder (PTLD) was not included as an SN. The final population for SIR analysis of SNs included 5933 patients (Figure 1).
Statistical analyses
The primary objectives of this study were to report the incidence of late mortality (defined as death in patients surviving at least 2 years posttransplantation) and the risk of SNs in children and adolescents receiving transplants for NMDs and assess whether those risks were increased relative to the general population of age- and sex-matched controls. Overall survival (OS) was calculated from 2 years after the date of transplantation until the date of death (or last contact for those who remained alive). Survival probabilities were estimated by the Kaplan-Meier method. Standardized mortality ratios (SMRs) with 95% confidence intervals (CIs) were used to compare observed mortality in the study population with expected mortality in the general population of age- and sex-matched controls in each country. Expected mortality was calculated from standardized actuarial tables in National Vital Statistics Reports6 and the Human Mortality Database.
In total, 86 pathology reports were requested from CIBMTR sites. Of these, 15 were removed because of misclassification of SNs. After review, 71 SN events were eligible for inclusion, 55 of which had enough accompanying information to be assigned an International Statistical Classification of Diseases and Related Health Problems, 10th edition (ICD-10), code. For analysis of SN incidence relative to the general population, the number of person-years at risk was calculated from the date of HCT until the date of last contact, SN diagnosis, or death. Incidence rates for all invasive cancers in the general population were obtained from selected registries worldwide.7-9 The risk of new malignancy in the study cohort was compared with that in the general population using in the method of Breslow et al,12 as described in previous CIBMTR studies.10-12 To compute expected numbers of cancers in the population, age, sex, calendar year, and region-specific incidence rates for all invasive solid cancers combined and for cancers of specific anatomic sites individually were applied to the appropriate person-years at risk.
All reported cases of SNs (with or without pathologic confirmation) that occurred after the date of HCT were treated as events (n = 71). Observed/expected ratios, also called SIRs, were calculated for each NMD group, and the exact Poisson distribution was used to calculate 95% CIs.10 For tumor site–specific SIR calculations, only cases to which we were able to assign an ICD-10 code were included (n = 55). A significance level α of 5% was used throughout.
Finally, multivariable Cox proportional hazards regression analyses were used to identify risk factors for SN development across the full patient cohort and in patients with FA or SAA and are presented as hazard ratios (HRs) with 95% CIs. Variables included in multivariable analyses were based on past research and known risk factors for SNs posttransplantation from prior studies. Models were adjusted for age, donor and stem cell source, myeloablative conditioning (vs reduced intensity or nonmyeloablative), receipt of total-body irradiation (TBI), and diagnosis of chronic GVHD. All analyses were performed with SAS software (version 9.4; SAS Institute, Cary, NC).
Results
Patient, disease, and transplantation characteristics
A summary of patient, disease, and transplantation characteristics is presented in Table 1. Median age at HCT was 6 years (interquartile range [IQR], 1-11; range, <1 to 20), and median follow-up was 7.8 years (IQR, 5.0-11.1; range, <1 to 20); 22% of the cohort had at least 10 years of follow-up. Forty percent of the cohort had congenital or acquired bone marrow failure disorders, including FA (10%) and SAA (24%). Patients with hemoglobinopathies comprised 15% of the cohort (SCD, 5%; thalassemia, 10%). Twenty-three percent of patients had either SCID or non-SCID PIDs, and 22% had inborn errors of metabolism, leukodystrophy, or osteopetrosis. A majority of patients received myeloablative conditioning (63%); however, only 17% of these regimens included TBI. A total of 71% of patients received antithymocyte globulin either as pretransplantation conditioning or as part of GVHD prophylaxis. Most patients received bone marrow as a stem cell source (65%). HLA-identical sibling (38%) or unrelated donors (17%) were most common (Table 1).
Patient and transplantation characteristics (N = 6028)
| Characteristic | n (%) |
|---|---|
| Follow-up of survivors, y | |
| Median | 7.8 |
| IQR | 5-11.1 |
| Range | <1 to 20 |
| Age at transplantation, y | |
| Median | 6 |
| IQR | 1-11 |
| Range | <1 to 20 |
| Sex | |
| Male | 3610 (60) |
| Female | 2418 (40) |
| Region of transplantation center | |
| United States | 3290 (55) |
| Canada | 195 (3) |
| Europe | 816 (13) |
| India and Asia | 307 (5) |
| Middle East and Africa | 697 (12) |
| Central and South America | 463 (8) |
| Australia and New Zealand | 251 (4) |
| Other | 9 (<1) |
| Year of transplantation | |
| 1995-1999 | 1763 (29) |
| 2000-2004 | 1803 (30) |
| 2005-2009 | 1977 (33) |
| 2010-2012 | 485 (8) |
| Primary disease category | |
| Bone marrow failure disorder | 2405 (40) |
| SAA | 1456 (24) |
| FA | 598 (10) |
| Other marrow failure | 351 (6) |
| Hemoglobinopathy | 874 (15) |
| Thalassemia | 574 (10) |
| SCD | 300 (5) |
| Immunodeficiency syndrome | 1415 (23) |
| SCID | 583 (10) |
| Non-SCID PID | 807 (13) |
| Histiocytic disorder | 469 (8) |
| Metabolic disease, leukodystrophy, other | 1334 (22) |
| Metabolic disorder | 476 (8) |
| Leukodystrophy | 227 (4) |
| Osteopetrosis | 150 (2) |
| Autoimmune disease | 25 (<1) |
| Other | 12 (<1) |
| Conditioning regimen | |
| None | 141 (2) |
| TBI + Cy ± other | 894 (15) |
| TBI + other | 117 (2) |
| Bu + Cy | 2720 (45) |
| Bu + other (not Cy) | 309 (5) |
| Cy + other (no Bu) | 1239 (21) |
| Fludarabine + other | 493 (8) |
| Other | 115 (2) |
| Graft type | |
| Bone marrow | 3922 (65) |
| Peripheral blood | 553 (9) |
| Cord blood | 1553 (26) |
| Donor | |
| HLA-identical sibling | 2298 (38) |
| Other related | 482 (8) |
| Unrelated | 1695 (28) |
| Cord blood | 1553 (26) |
| Characteristic | n (%) |
|---|---|
| Follow-up of survivors, y | |
| Median | 7.8 |
| IQR | 5-11.1 |
| Range | <1 to 20 |
| Age at transplantation, y | |
| Median | 6 |
| IQR | 1-11 |
| Range | <1 to 20 |
| Sex | |
| Male | 3610 (60) |
| Female | 2418 (40) |
| Region of transplantation center | |
| United States | 3290 (55) |
| Canada | 195 (3) |
| Europe | 816 (13) |
| India and Asia | 307 (5) |
| Middle East and Africa | 697 (12) |
| Central and South America | 463 (8) |
| Australia and New Zealand | 251 (4) |
| Other | 9 (<1) |
| Year of transplantation | |
| 1995-1999 | 1763 (29) |
| 2000-2004 | 1803 (30) |
| 2005-2009 | 1977 (33) |
| 2010-2012 | 485 (8) |
| Primary disease category | |
| Bone marrow failure disorder | 2405 (40) |
| SAA | 1456 (24) |
| FA | 598 (10) |
| Other marrow failure | 351 (6) |
| Hemoglobinopathy | 874 (15) |
| Thalassemia | 574 (10) |
| SCD | 300 (5) |
| Immunodeficiency syndrome | 1415 (23) |
| SCID | 583 (10) |
| Non-SCID PID | 807 (13) |
| Histiocytic disorder | 469 (8) |
| Metabolic disease, leukodystrophy, other | 1334 (22) |
| Metabolic disorder | 476 (8) |
| Leukodystrophy | 227 (4) |
| Osteopetrosis | 150 (2) |
| Autoimmune disease | 25 (<1) |
| Other | 12 (<1) |
| Conditioning regimen | |
| None | 141 (2) |
| TBI + Cy ± other | 894 (15) |
| TBI + other | 117 (2) |
| Bu + Cy | 2720 (45) |
| Bu + other (not Cy) | 309 (5) |
| Cy + other (no Bu) | 1239 (21) |
| Fludarabine + other | 493 (8) |
| Other | 115 (2) |
| Graft type | |
| Bone marrow | 3922 (65) |
| Peripheral blood | 553 (9) |
| Cord blood | 1553 (26) |
| Donor | |
| HLA-identical sibling | 2298 (38) |
| Other related | 482 (8) |
| Unrelated | 1695 (28) |
| Cord blood | 1553 (26) |
Bu, busulfan; Cy, cyclophosphamide; SCID, severe combined immunodeficiency.
OS and relative mortality among 2-year survivors
A total of 4214 patients (70%) with at least 2 years of follow-up were included in analysis of late mortality; 1524 (25%) died before 2 years, and 251 (4%) were excluded because of incomplete data at 2 years posttransplantation. Of the 25% of patients who died before 2 years, the primary causes of death were organ failure (23%) and infection (22%), followed by pulmonary complications (12%) and GVHD (10%; Table 2). OS probabilities among the patients still alive after 2 years were 97% (95% CI, 96% to 97%) and 93% (95% CI, 92% to 94%) at 5 and 10 years, respectively (Table 3). Long-term survival in this cohort varied based on underlying NMD. Patients with inborn errors of metabolism or leukodystrophies had the lowest survival probabilities at each evaluable time point (Table 3). The most common cause of death in 2-year survivors was organ failure (20%), followed by complications of primary disease (17%), infection (11%), and GVHD (10%). Subsequent neoplasms were the cause of death in 6% of cases (Table 2).
Cause of death <2 vs ≥2 years posttransplantation (N = 6028)
| Primary cause of death | <2 y, n (%) | ≥2 y, n (%) | Total, n (%) |
|---|---|---|---|
| Patients, n | 1524 | 251 | 1775 |
| Primary disease | 104 (7) | 43 (17) | 147 (8) |
| Graft failure | 87 (6) | 9 (4) | 96 (5) |
| GVHD | 151 (10) | 26 (10) | 177 (10) |
| Infection | 332 (22) | 28 (11) | 360 (20) |
| Pulmonary/ARDS | 182 (12) | 11 (4) | 193 (11) |
| Organ failure | 348 (23) | 50 (20) | 398 (22) |
| Secondary malignancy | 19 (1) | 14 (6) | 33 (2) |
| Other cause | 147 (10) | 33 (13) | 180 (10) |
| Unknown | 2 (0) | 0 (0) | 2 (0) |
| Missing | 152 (10) | 37 (15) | 189 (11) |
| Primary cause of death | <2 y, n (%) | ≥2 y, n (%) | Total, n (%) |
|---|---|---|---|
| Patients, n | 1524 | 251 | 1775 |
| Primary disease | 104 (7) | 43 (17) | 147 (8) |
| Graft failure | 87 (6) | 9 (4) | 96 (5) |
| GVHD | 151 (10) | 26 (10) | 177 (10) |
| Infection | 332 (22) | 28 (11) | 360 (20) |
| Pulmonary/ARDS | 182 (12) | 11 (4) | 193 (11) |
| Organ failure | 348 (23) | 50 (20) | 398 (22) |
| Secondary malignancy | 19 (1) | 14 (6) | 33 (2) |
| Other cause | 147 (10) | 33 (13) | 180 (10) |
| Unknown | 2 (0) | 0 (0) | 2 (0) |
| Missing | 152 (10) | 37 (15) | 189 (11) |
ARDS, acute respiratory distress syndrome.
Survival probabilities at 5 and 10 years in patients alive ≥2 years post-HCT, with corresponding SMRs, by NMD
| Primary disease | n (%) | 5 y (95% CI) | 10 y (95% CI) | SMR (95% CI) | P |
|---|---|---|---|---|---|
| Patients alive ≥2 y post-HCT | n = 4214 | 97 (96-97) | 93 (92-94) | 4.2 (3.7-4.8) | <.0001 |
| Bone marrow failure disorder | |||||
| SAA | 192 (5) | 98 (97-99) | 95 (93-97) | 1.8 (1.3-2.4) | .0009 |
| FA | 393 (9) | 96 (94-98) | 92 (88-95) | 4.8 (3.4-6.6) | <.0001 |
| Other marrow failure | 242 (6) | 95 (91-97) | 93 (88-96) | 6.4 (3.8-10) | <.0001 |
| Hemoglobinopathy | |||||
| Thalassemia | 401 (10) | 98 (97-99) | 98 (97-99) | 0.7 (0.3-1.4) | .3942 |
| SCD | 247 (6) | 97 (94-99) | 96 (93-98) | 8.3 (3.9-15) | <.0001 |
| Immunodeficiency syndrome | |||||
| SCID | 403 (10) | 97 (95-98) | 93 (90-96) | 4.8 (3.0-7.3) | <.0001 |
| Non-SCID PID | 607 (14) | 97 (96-99) | 95 (93-97) | 5.0 (3.3-7.4) | <.0001 |
| Histiocytic disorder | 273 (6) | 98 (96-99) | 94 (89-97) | 9.3 (4.8-16) | <.0001 |
| Inborn error of metabolism, leukodystrophy, other | |||||
| Metabolic disorder | 314 (7) | 93 (89-95) | 86 (81-90) | 25 (18-5) | <.0001 |
| Leukodystrophy | 143 (3) | 89 (83-94) | 75 (64-84) | 37 (37-53) | <.0001 |
| Osteopetrosis | 77 (2) | 93 (86-98) | 85 (74-93) | 11.5 (5.5-21) | <.0001 |
| Autoimmune disease | 12 (<1) | 100 | 83 (46-100) | 12.9 (0.3-72) | .1498 |
| Primary disease | n (%) | 5 y (95% CI) | 10 y (95% CI) | SMR (95% CI) | P |
|---|---|---|---|---|---|
| Patients alive ≥2 y post-HCT | n = 4214 | 97 (96-97) | 93 (92-94) | 4.2 (3.7-4.8) | <.0001 |
| Bone marrow failure disorder | |||||
| SAA | 192 (5) | 98 (97-99) | 95 (93-97) | 1.8 (1.3-2.4) | .0009 |
| FA | 393 (9) | 96 (94-98) | 92 (88-95) | 4.8 (3.4-6.6) | <.0001 |
| Other marrow failure | 242 (6) | 95 (91-97) | 93 (88-96) | 6.4 (3.8-10) | <.0001 |
| Hemoglobinopathy | |||||
| Thalassemia | 401 (10) | 98 (97-99) | 98 (97-99) | 0.7 (0.3-1.4) | .3942 |
| SCD | 247 (6) | 97 (94-99) | 96 (93-98) | 8.3 (3.9-15) | <.0001 |
| Immunodeficiency syndrome | |||||
| SCID | 403 (10) | 97 (95-98) | 93 (90-96) | 4.8 (3.0-7.3) | <.0001 |
| Non-SCID PID | 607 (14) | 97 (96-99) | 95 (93-97) | 5.0 (3.3-7.4) | <.0001 |
| Histiocytic disorder | 273 (6) | 98 (96-99) | 94 (89-97) | 9.3 (4.8-16) | <.0001 |
| Inborn error of metabolism, leukodystrophy, other | |||||
| Metabolic disorder | 314 (7) | 93 (89-95) | 86 (81-90) | 25 (18-5) | <.0001 |
| Leukodystrophy | 143 (3) | 89 (83-94) | 75 (64-84) | 37 (37-53) | <.0001 |
| Osteopetrosis | 77 (2) | 93 (86-98) | 85 (74-93) | 11.5 (5.5-21) | <.0001 |
| Autoimmune disease | 12 (<1) | 100 | 83 (46-100) | 12.9 (0.3-72) | .1498 |
Comparisons of mortality rates in each disease group with rates in the general population are presented in Table 2. Although long-term survival was excellent, the study cohort alive at 2 years still had a fourfold increased incidence of all-cause mortality compared with age- and sex-matched controls from the general population (SMR, 4.2; 95% CI, 3.7-4.8). Mortality rates for all NMDs were significantly higher than rates in the general population, with the exception of patients with thalassemia or autoimmune disease. Increased mortality rate was most dramatically noted in children with inborn errors of metabolism or leukodystrophies, whose risks of death were 37- and 25-fold higher than expected, respectively (Table 2).
SNs
Among 5933 patients with median follow-up of 7.6 years (range, 2-20.9), 71 (1.2%) developed SNs. Details of the 71 individual cases are presented in supplemental Table 1. Incidence of SNs was highest in children with FA (5.5%), SAA (1.1%), or other marrow failure disorders (1.7%; Table 4). In patients with hemoglobinopathies, PIDs, metabolic disorders, leukodystrophies, or other NMDs, SN rates were <1%. No SNs were observed in patients receiving transplants for thalassemia. The most frequent SNs were hematologic malignancies, including acute myeloid leukemia (AML; n = 8), myelodysplastic syndrome (MDS; n = 7), and acute lymphoblastic leukemia (n = 3). Oropharyngeal cancers, including mouth (n = 2), tongue (n = 5), and hypopharynx (n = 1), comprised 25% of cases. Skin cancers, including melanoma (n = 2), were diagnosed in 9 patients (Table 4).
Incidence of SNs by primary NMD with SIRs (N = 5933)
| Primary NMD | n of survivors (median follow-up, y; range) | Reported SNs, n (%) | SIR (95% CI) | P |
|---|---|---|---|---|
| Total | 5933 (7.6; 0-20.9) | 71 (1.2) | 11 (9-14) | <.001 |
| Bone marrow failure disorder | ||||
| SAA | 1429 (7.6; 0-20.8) | 16 (1.1) | 8 (4-12) | <.001 |
| FA | 574 (9.1; 0.2-20.4) | 31 (5.4) | 50 (34-71) | <.001 |
| Other marrow failure | 343 (7.7; 0.6-20.3) | 6 (1.7) | 15 (5-32) | <.001 |
| Hemoglobinopathy | ||||
| SCD | 294 (6.1; 0.1-20.1) | 3 (1) | 11 (2-33) | .005 |
| Thalassemia | 572 (5.3; 0.2-20.0) | 0 (0) | — | — |
| Immunodeficiency syndrome | ||||
| SCID | 575 (7.3; 0.2-20.9) | 2 (0.3) | 4 (0.5-14) | .196 |
| Non-SCID PID | 793 (8.0; 0.2-20.5) | 3 (0.4) | 4 (0.7-10) | .107 |
| Histiocytic disorder | 466 (7.1; 0.5-20.2) | 5 (1) | 14 (5-33) | <.001 |
| Metabolic disease, leukodystrophy, other | ||||
| Leukodystrophy | 227 (6.1; 0.2-20.2) | 2 (0.9) | 10 (1-37) | .034 |
| Osteopetrosis | 150 (6.9; 0.2-20.2) | 1 (0.7) | 10 (0.3-56) | .191 |
| Metabolic disorder | 474 (8.3; 0.2-20.4) | 2 (0.4) | 4 (0.5-15) | .172 |
| Autoimmune | 24 (4.7; 0.3-15.8) | 0 (0) | — | — |
| Other | 12 (8.7; 5.5-14.0) | 0 (0) | — | — |
| Primary NMD | n of survivors (median follow-up, y; range) | Reported SNs, n (%) | SIR (95% CI) | P |
|---|---|---|---|---|
| Total | 5933 (7.6; 0-20.9) | 71 (1.2) | 11 (9-14) | <.001 |
| Bone marrow failure disorder | ||||
| SAA | 1429 (7.6; 0-20.8) | 16 (1.1) | 8 (4-12) | <.001 |
| FA | 574 (9.1; 0.2-20.4) | 31 (5.4) | 50 (34-71) | <.001 |
| Other marrow failure | 343 (7.7; 0.6-20.3) | 6 (1.7) | 15 (5-32) | <.001 |
| Hemoglobinopathy | ||||
| SCD | 294 (6.1; 0.1-20.1) | 3 (1) | 11 (2-33) | .005 |
| Thalassemia | 572 (5.3; 0.2-20.0) | 0 (0) | — | — |
| Immunodeficiency syndrome | ||||
| SCID | 575 (7.3; 0.2-20.9) | 2 (0.3) | 4 (0.5-14) | .196 |
| Non-SCID PID | 793 (8.0; 0.2-20.5) | 3 (0.4) | 4 (0.7-10) | .107 |
| Histiocytic disorder | 466 (7.1; 0.5-20.2) | 5 (1) | 14 (5-33) | <.001 |
| Metabolic disease, leukodystrophy, other | ||||
| Leukodystrophy | 227 (6.1; 0.2-20.2) | 2 (0.9) | 10 (1-37) | .034 |
| Osteopetrosis | 150 (6.9; 0.2-20.2) | 1 (0.7) | 10 (0.3-56) | .191 |
| Metabolic disorder | 474 (8.3; 0.2-20.4) | 2 (0.4) | 4 (0.5-15) | .172 |
| Autoimmune | 24 (4.7; 0.3-15.8) | 0 (0) | — | — |
| Other | 12 (8.7; 5.5-14.0) | 0 (0) | — | — |
The observed incidence of SNs posttransplantation was significantly higher than the expected incidence in the general population (SIR, 11; 95% CI, 8.9-13.9; Table 5). Excess risk was evident in those receiving transplants for FA (SIR, 50; 95% CI, 34-71), SAA (SIR, 8; 95% CI, 4-12), or SCD (SIR, 11; 95% CI, 2-33). Patients with leukodystrophy or histiocytic disorders had 10- (95% CI, 1-37) and 14-fold (95% CI, 5-33) higher-than-expected incidence of SNs, respectively. Posttransplantation SN incidence was not significantly higher than expected in patients undergoing transplantation for thalassemia, PIDs, osteopetrosis, or metabolic disorders (Table 5).
Site-specific tumor incidence with SIRs (N = 5933; 35 667 person-years)
| Tumor site | Reported SN cases, n (%) or n | SIR (95% CI)* | P |
|---|---|---|---|
| Head and neck | 18 (25) | ||
| Tongue | 5 | 490 (159 to >1000) | <.001 |
| Mouth | 2 | 113 (14-408) | <.001 |
| Oropharynx | 2 | >1000 (309 to >1000) | <.0001 |
| Hypopharynx | 1 | >1000 (32 to >1000) | .002 |
| Nose, sinuses | 1 | 47 (1.2-264) | .042 |
| Oropharynx, unspecified | 7 | — | — |
| Hematologic | 19 (27) | ||
| Lymphoid leukemia | 3 | 3 (0.65-9) | .143 |
| Myeloid leukemia | 8 | 23 (9.7-44) | <.001 |
| MDS | 7 | 730 (293 to >1000) | <.001 |
| MDS, unspecified | 1 | — | — |
| Skin | 9 (13) | ||
| Melanoma of the skin | 2 | 7 (0.8-25) | .071 |
| Nonmelanoma skin | 7 | 68 (27-140) | <.001 |
| Bone and soft tissue | 7 (10) | ||
| Connective, soft tissue | 2 | 6 (0.8-23) | .083 |
| Bone | 2 | 6 (0.7-21) | .096 |
| Sarcoma, unspecified | 3 | — | — |
| Thyroid | 5 (7) | 16 (5.2-37) | <.001 |
| GI/GU | 7 (10) | ||
| Liver | 2 | 28 (3-100) | .005 |
| Cervix uteri | 1 | 13 (0.3-74) | .145 |
| Testis | 1 | 3 (0.1-15) | .607 |
| Bladder | 1 | 42 (1.0-234) | .047 |
| GI/GU, unspecified | 2 | — | — |
| Brain, CNS | 2 (3) | 2 (0.3-8) | .473 |
| Breast | 1 (1) | 10 (0.3-58) | .184 |
| Other, unspecified | 3 (4) | — | — |
| Tumor site | Reported SN cases, n (%) or n | SIR (95% CI)* | P |
|---|---|---|---|
| Head and neck | 18 (25) | ||
| Tongue | 5 | 490 (159 to >1000) | <.001 |
| Mouth | 2 | 113 (14-408) | <.001 |
| Oropharynx | 2 | >1000 (309 to >1000) | <.0001 |
| Hypopharynx | 1 | >1000 (32 to >1000) | .002 |
| Nose, sinuses | 1 | 47 (1.2-264) | .042 |
| Oropharynx, unspecified | 7 | — | — |
| Hematologic | 19 (27) | ||
| Lymphoid leukemia | 3 | 3 (0.65-9) | .143 |
| Myeloid leukemia | 8 | 23 (9.7-44) | <.001 |
| MDS | 7 | 730 (293 to >1000) | <.001 |
| MDS, unspecified | 1 | — | — |
| Skin | 9 (13) | ||
| Melanoma of the skin | 2 | 7 (0.8-25) | .071 |
| Nonmelanoma skin | 7 | 68 (27-140) | <.001 |
| Bone and soft tissue | 7 (10) | ||
| Connective, soft tissue | 2 | 6 (0.8-23) | .083 |
| Bone | 2 | 6 (0.7-21) | .096 |
| Sarcoma, unspecified | 3 | — | — |
| Thyroid | 5 (7) | 16 (5.2-37) | <.001 |
| GI/GU | 7 (10) | ||
| Liver | 2 | 28 (3-100) | .005 |
| Cervix uteri | 1 | 13 (0.3-74) | .145 |
| Testis | 1 | 3 (0.1-15) | .607 |
| Bladder | 1 | 42 (1.0-234) | .047 |
| GI/GU, unspecified | 2 | — | — |
| Brain, CNS | 2 (3) | 2 (0.3-8) | .473 |
| Breast | 1 (1) | 10 (0.3-58) | .184 |
| Other, unspecified | 3 (4) | — | — |
CNS, central nervous system; GI, gastrointestinal; GU, genitourinary.
Based on number of SN cases with assigned ICD-10 codes (n = 55).
Tumor site–specific comparisons of SNs in the study cohort with the general population were restricted to the 55 cases assigned ICD-10 codes. Analysis revealed that risk in the study cohort was elevated for almost all cancer sites (Table 5). Compared with the general population, the study cohort had significantly increased rates of AML (SIR, 23; 95% CI, 9.7-44) and MDS (SIR, 730; 95% CI, 293 to >1000), as well as increased rates of oropharyngeal, skin, thyroid, liver, and bladder tumors. Of particular note were the higher-than-expected rates of tongue (SIR, 490; 95% CI, 159 to >1000) and mouth tumors (SIR, 113; 95% CI, 14-408), which developed exclusively in patients receiving transplants for FA or marrow failure syndromes. Patients in the study cohort also had higher-than-expected rates of nonmelanomatous skin cancer (SIR, 68; 95% CI, 27-140). The incidence of bone cancer and central nervous system tumors was not significantly higher in the study cohort than would be expected in the general population.
In the full cohort of patients, multivariable models revealed that TBI-based conditioning was associated with a significantly higher hazard of SNs. Specifically, patients who received a TBI-based conditioning regimen had a 2.5-fold higher hazard of developing a posttransplantation SN compared with those who did not receive TBI-based conditioning (HR, 2.5; 95% CI, 1.5-4.1; P = .0003). Among patients with FA, nonmyeloablative conditioning was associated with a 75% reduction in the hazard of SNs (HR, 0.26; 95% CI, 0.1-0.9; P = .0271). Development of chronic GVHD in patients with FA was associated with a 4.8-fold increased hazard of SN development (HR, 4.8; 95% CI, 2.3-10.4; P <.0001).
Median time from HCT to SN development was 7.5 years (IQR, 3.5-10.4; range, 0.04-18.5; Figure 2). Median latency between transplantation and SN was shortest for AML (median, 1.8 years; IQR, 1.0-3.7). Latency was longer for cancers of the oropharynx (median, 10.2 years; IQR, 8.2-14.5), as well as for skin (median, 7.2 years; IQR, 2.9-7.9) and other solid tumors (median, 8.6 years; IQR, 6.1-10.6; Figure 2). Information on 71 individual SN cases with details about primary NMD, pretransplantation conditioning, TBI, and GVHD is presented in supplemental Table 1.
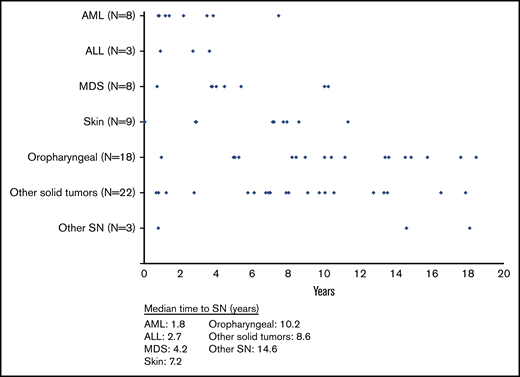
Median time to SN development after HCT. Median time to SNs for all new diagnoses was 7.5 years (IQR, 3.5-10.4; range, <1 to 18.5). Median time to SNs by tumor type was as follows: AML: 1.8 years (IQR, 1.0-3.7; range, <1 to 7.5); acute lymphoblastic leukemia (ALL): 2.7 years (IQR, <1 to 3.6; range, <1 to 3.6); MDS: 4.2 years (IQR, 3.8-7.7; range, <1 to 10.2); skin: 7.2 years (IQR, 2.9-7.9; range, <1 to 11.3); oropharyngeal: 10.2 years (IQR, 8.2-14.5; range, 1-18.5); solid tumors: 8.6 years (IQR, 6.1-10.6; range, <1 to 17.9); and other SNs, unspecified: 14.6 years (IQR, <1 to 18.1; range, <1 to 18.1).
Median time to SN development after HCT. Median time to SNs for all new diagnoses was 7.5 years (IQR, 3.5-10.4; range, <1 to 18.5). Median time to SNs by tumor type was as follows: AML: 1.8 years (IQR, 1.0-3.7; range, <1 to 7.5); acute lymphoblastic leukemia (ALL): 2.7 years (IQR, <1 to 3.6; range, <1 to 3.6); MDS: 4.2 years (IQR, 3.8-7.7; range, <1 to 10.2); skin: 7.2 years (IQR, 2.9-7.9; range, <1 to 11.3); oropharyngeal: 10.2 years (IQR, 8.2-14.5; range, 1-18.5); solid tumors: 8.6 years (IQR, 6.1-10.6; range, <1 to 17.9); and other SNs, unspecified: 14.6 years (IQR, <1 to 18.1; range, <1 to 18.1).
Discussion
To our knowledge, this is the largest study examining SN incidence and late mortality in an international cohort of children and adolescents receiving transplants for NMDs. Findings indicate that a majority of patients who undergo HCT for treatment of NMDs and are still alive after 2 years have excellent long-term survival. These findings, while encouraging, underscore the fact that despite low cumulative incidence of late mortality, the risk of early death remains a significant challenge in the pediatric transplantation population. Furthermore, the risk of death resulting from GVHD, infection, or organ failure persists over time, particularly in those undergoing transplantation for inborn errors of metabolism or leukodystrophies. Despite our cohort having higher-than-expected mortality when compared with the general population, a majority of patients who remained alive 2 years posttransplantation had high survival rates at both 5 and 10 years. With the exception of patients undergoing transplantation for FA, the study cohort had a reassuringly low cumulative incidence of posttransplantation SNs.
The cumulative incidence of mortality in our long-term survivors is similar to other recent reports of patients undergoing allogeneic HCT for NMDs. In a recent study examining posttransplantation mortality in patients still alive at 2 years, the 20-year OS rate by primary diagnosis was 92% for immune disorders, 91% for SAA, 82.3% for sickle cell anemia or thalassemia, and 68.5% for FA. When compared with the general population, the relative mortality in patients undergoing transplantation between 2000 and 2010 was actually higher than in earlier time periods. The authors suggested this could be related to the relatively lower rate of childhood death resulting from other causes. Overall, they noted that the cumulative incidence of late mortality decreased significantly over time from 1990 to 2010.13 In 2007, Bhatia et al14 reported 5- and 10-year survival outcomes in patients with SAA of 96% and 94%, respectively.
For children in our cohort who received transplants for FA, SAA, or marrow failure, 10-year survival probabilities were >90%. In our analysis, which was restricted to patients still alive at 2 years, survival probabilities at 5 and 10 years for patients with SAA were 98% and 95%, respectively. These rates are substantially higher than earlier reports of outcomes in SAA,15 which may be related to the younger age of our cohort or continued improvements in the transplantation procedure, as has been suggested in other recent reports. It should be noted that the present analysis, in contrast to prior studies, did not include 1-year transplantation-related mortality, which may also have contributed to the relatively improved outcomes. Similar to recent studies reporting survival outcomes near 100% in children receiving matched sibling donor transplants for SCD, we observed excellent long-term survival in this group, with 5- and 10-year survival probabilities of 97% and 96%, respectively.16 We observed no excess risk of death in patients receiving transplants for thalassemia. In 2012, Bernardo et al17 reported 5-year survival of 93% in younger patients undergoing transplantation for thalassemia, which is closer to the probabilities observed in our cohort.
We observed favorable long-term survival in patients with PIDs, which has been reported by others, although inconsistently.3,18 Pai et al19 reported that in patients who receive transplants before the onset of infection, excellent survival is expected in infants with SCID. Survival in our cohort was similar to that of the younger patients.20 Again, differences in mortality risk observed between our study and earlier studies may in part be attributed to differences in cohort selection criteria (1- vs 2-year survivors). Few studies have reported long-term outcomes in patients undergoing transplantation for histiocytic disorders. We observed that survival for these patients was excellent at both 5 and 10 years (98% and 94%, respectively).
Ten-year survival probabilities in patients with inborn errors of metabolism or leukodystrophies ranged from 75% to 86%. In an analysis of OS in children undergoing cord blood transplantation for leukodystrophies, Van den Broek et al21 reported similar survival probabilities to those observed in our cohort despite not restricting their study to 2-year survivors. Survival probabilities were similar to what we observed in our cohort, suggesting that the risk of posttransplantation mortality, which is often highest in the early posttransplantation period, may persist over time in this patient population.
Overall, the study cohort had a low incidence of posttransplantation SNs (1.2%). With median follow-up of nearly 8 years, SN incidence was 5.5% in patients with FA and 1.1% in patients with SAA.22 These rates are slightly lower than those of prior reports in this patient population with similar length of follow-up.23 In one of the earliest studies examining posttransplantation SN rates, Socié et al24 reported an 8-year SN incidence up to 22% in children receiving transplants for SAA. More recent studies with longer follow-up have reported higher rates of SNs, which may reflect the long latency between transplantation and SN development. In 2014, the European Group for Blood and Marrow Transplantation reported that cumulative incidences of SNs after transplantation in patients with FA were 21% at 15 years and 34% at 20 years post-HCT.25
Our observation that patients with FA had an increased SN risk compared with the general population is consistent with other studies of FA patients. Even in the absence of HCT, the underlying genetic instability associated with FA renders these patients at increased risk for head and neck tumors, as well as for hematologic and solid malignancies.26 In patients with FA who undergo HCT, exposure to TBI or alkylating agents, immunosuppression, and GVHD development have been associated with SN development. We observed that chronic GVHD was a significant risk factor for SN development in patients with FA. The use of non–TBI-based conditioning regimens is increasingly common in recent decades. Whether this practice shift will reduce the rates of SNs in this population remains to be seen.
Rates of posttransplantation SNs were low in patients undergoing transplantation for SCD or thalassemia. Studies in patients with thalassemia have reported that rates of posttransplantation SNs are similar to rates in patients who do not undergo HCT.27 In our cohort, we did not observe any SNs in patients with thalassemia. Recently, Keegan et al28 reported a higher incidence of leukemia in patients with SCD compared with the general population. However, in our cohort, SN risk after transplantation for SCD was not increased compared with the general population. It is plausible that treatment of SCD, a disease that is confined exclusively to the hematopoietic system, may have reduced the likelihood of developing MDS or leukemia later on.
Patients with PIDs also had low rates of posttransplantation SNs. Historically, patients with PIDs may be at a higher risk of developing PTLD in the early post-HCT period. In our study, the incidence of SNs in patients receiving transplants for primary immunodeficiencies was <1%; however, PTLD was not included as an SN for the purposes of the present study. Patients with histiocytic disorders had an SN rate of 1.1%, which was significantly higher than expected in the general population. Oropharyngeal, mucosal, and nonmelanomatous skin cancers were the most common nonhematologic SNs observed in our cohort. All patients with oropharyngeal cancers had an underlying diagnosis of FA (supplemental Table 1). Timing of SN development in our cohort was similar to that in other reports.29 In addition to early leukemia and skin cancer, we observed a long-term risk for thyroid, oropharyngeal, and liver tumors, as well as a persistent risk for MDS. A majority of nonleukemic SNs occurred ≥5 years post-HCT.
A strength of this study is the large, international cohort of children and adolescents, with detailed treatment information and long duration of follow-up. The analysis builds on earlier work from the CIBMTR, which examined mortality rates in long-term survivors after HCT for primary malignancy or SAA before 2004.14,30 The present analysis extends this work, with 8 more years of data in a cohort with a broader spectrum of NMDs. Similar to other reports, we found that TBI is still associated with a higher hazard of SNs, and early posttransplantation mortality rates remain unacceptably high. We were unable to evaluate pretransplantation organ dysfunction related to underlying disease, which could have contributed to survival outcomes, particularly in patients with metabolic syndromes or leukodystrophies. For data collected through the CIBMTR, there is central pathology review for SN cases, and we were unable to obtain enough data to assign ICD-10 codes for 16 of the reported cases. These SNs were not included in the analysis of relative incidence; therefore, the true SIR for some NMD groups and some site-specific cancer diagnoses may have been underestimated. Long-term follow-up beyond 10 years was only available in a subset of our cohort, which also may have led to underestimation of SN risk, because many malignancies often develop beyond 15 years posttransplantation.27 Additional follow-up will be necessary to determine longer-term survival and later SN development in this patient cohort.
In summary, we report excellent long-term survival in patients still alive at 2 years, as well as low incidence of posttransplantation SNs, in an international cohort of children and adolescents undergoing transplantation for NMDs. These findings are accompanied by a recognition that early posttransplantation mortality remains a significant challenge and that the risk for SN development persists over time. The patients at highest risk of developing SNs were those who might have had underlying predispositions to malignancy, specifically patients with FA or marrow failure disorders. In the full cohort, TBI-based conditioning regimens were associated with a significantly higher hazard of SN development; in patients with FA, the hazard of SNs was significantly higher in those with chronic GVHD. We observed no excess cancer risk in children receiving transplants for thalassemia, PIDs, osteopetrosis, and metabolic disorders. The present findings suggest that in the absence of individual patient susceptibility, the transplantation procedure itself may not necessarily increase the likelihood of developing a malignancy. It is important to note that a majority of nonleukemic SNs developed at least 5 years posttransplantation, which highlights the need for long-term surveillance and close follow-up in children and adolescents undergoing transplantation for NMDs.31
Data Sharing requests should be e-mailed to the corresponding author, Rachel Phelan (rphelan@mcw.edu).
Acknowledgments
The Center for International Blood and Marrow Transplant Research is supported primarily by Public Health Service U24CA076518 from the National Institutes of Health (NIH), National Cancer Institute (NCI), the National Heart, Lung and Blood Institute (NHLBI) and the National Institute of Allergy and Infectious Diseases (NIAID); U24HL138660 from NIH, NHLBI and NCI; R21HL140314 and U01HL128568 from the NIH, NHLBI; HHSH250201700006C, SC1MC31881-01-00 and HHSH250201700007C from the Health Resources and Services Administration; and N00014-18-1-2850, N00014-18-1-2888, and N00014-20-1-2705 from the Office of Naval Research. Additional federal support is provided by NIH, NCI P01CA111412, R01CA152108, R01CA215134, R01CA218285, and R01CA231141; NIH, NHLBI R01HL126589, R01HL129472, R01HL130388, and R01HL131731; NIH, NIAID R01AI128775, U01AI069197, and U01AI126612; and Biomedical Advanced Research and Development Authority. Support is also provided by Be the Match Foundation, Boston Children’s Hospital, Dana-Farber, Japan Hematopoietic Cell Transplantation Data Center, St. Baldrick’s Foundation, the National Marrow Donor Program, the Medical College of Wisconsin, and from the following commercial entities: AbbVie; Actinium Pharmaceuticals, Inc.; Adaptive Biotechnologies; Adienne SA; Allovir, Inc.; Amgen, Inc.; Anthem, Inc.; Astellas Pharma US; AstraZeneca; Atara Biotherapeutics, Inc.; bluebird bio, Inc.; Bristol-Myers Squibb Co.; Celgene Corp.; Chimerix, Inc.; CSL Behring; CytoSen Therapeutics, Inc.; Daiichi Sankyo Co., Ltd.; Gamida-Cell, Ltd.; Genzyme; GlaxoSmithKline; HistoGenetics, Inc.; Incyte Corporation; Janssen Biotech, Inc.; Janssen Pharmaceuticals, Inc.; Janssen/Johnson & Johnson; Jazz Pharmaceuticals, Inc.; Kiadis Pharma; Kite Pharma; Kyowa Kirin; Legend Biotech; Magenta Therapeutics; Mallinckrodt LLC; Medac GmbH; Merck & Company, Inc.; Merck Sharp & Dohme Corp.; Mesoblast; Millennium, Takeda Oncology Co.; Miltenyi Biotec, Inc.; Novartis Oncology; Novartis Pharmaceuticals Corporation; Omeros Corporation; Oncoimmune, Inc.; Orca Biosystems, Inc.; Pfizer, Inc.; Phamacyclics, LLC; Regeneron Pharmaceuticals, Inc.; REGiMMUNE Corp.; Sanofi Genzyme; Seattle Genetics; Sobi, Inc.; Takeda Oncology; Takeda Pharma; Terumo BCT; Viracor Eurofins; and Xenikos BV.
The views expressed in this article do not reflect the official policy or position of the NIH, the US Department of the Navy, the US Department of Defense, the Health Resources and Services Administration, or any other agency of the US government.
Authorship
Contribution: J.M.K., P.S., and B.E.S. designed, directed, and performed research, analyzed data, and wrote the manuscript; H.R.T., R.P., and S.B.-S. provided data sets for analysis; H.R.T., S.B.-S., and R.B. performed statistical analysis; B.E.S., R.P., D.B., M.B., M.E.D.F., and B.N.S. reviewed data and the manuscript; and L.B., A.A.A., A.K.K., A.D., B.W., B.G., B.P.A., C.U., C.O.F., A.M.B., C.D., E.C., G.C.H., H.S.M., H.M.L., J.J.A., K.C.M., K.M.W., K.M.P., L.M.V., M.N., M.B., M.A.D., N.K., N.S.B., A.R., N.F., P.A.M., P.H., P.J.S., R.T.K., R.S., R.F.O., R.J.H., R.P.G., S.J.M., S.C., S.J.R., S.M.B., S.G., S.P., T.N., T.P., V.A., W.J.H., and Y.I. critically reviewed the data and approved the final manuscript before it was submitted.
Conflict-of-interest disclosure: P.J.S. is on the steering committee for Pharmacyclics: Ibrutinib. R.F.O. is a consultant and stockholder with AstraZeneca. S.G. is a speaker with Seattle Genetics and Kite Pharma and has participated in advisory boards with Janssen, Amgen, and Kadmon. A.R. was a member of one-time ad hoc scientific advisory boards for Nohla Therapeutics and Kaleido; has served as a medical expert witness for the US Department of Justice; and his brother is employed by Johnson & Johnson. G.C.H. has stock and ownership in Sangamo Bioscience, CVS Health, Juno Theraputics, Celgene, Kite Pharma, Bluebird Bio, Novartis, Bristol-Myers Squibb/Medarex, Insys Therapeutics, Crispr Therapeutics, AbbVie, IDEXX Laboratories, GW Pharmaceuticals, Johnson & Johnson, Cardinal Health, Pfizer, Immunomedics, Procter & Gamble, Endocyte, Vertex, Clovis Oncology, Jazz Pharmaceuticals, Cellectis, and Aetna; is a consultant with Pfizer, Kite Pharma, Incyte, and Jazz Pharmaceuticals; reports research funding with Takeda, Jazz Pharmaceuticals, and Pharmacyclics; and travel and accommodations with Kite Pharma, Incyte, Pfizer, Falk Foundation, Jazz Pharmaceuticals, and Astellas Pharma. R.P. participated on a one-time advisory board for Orchard Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Rachel Phelan, Medical College of Wisconsin, 8701 Watertown Plank Rd, PO Box 26509, Milwaukee, WI 53226; e-mail: rphelan@mcw.edu.

