

Key Points
IFNβ can be coadministered with tPA to alleviate delayed tPA–aggravated BBB disruption and HT in ischemic stroke.
IFNβ-mediated alleviation of delayed tPA–induced adverse effects is attributed to its effects on the suppression of MMP3/9 production.

Abstract
Tissue plasminogen activator (tPA) is the only US Food and Drug Administration (FDA)–approved drug for ischemic stroke. However, delayed tPA administration is associated with increased risk of blood-brain barrier (BBB) disruption and hemorrhagic transformation (HT). Interferon-β (IFNβ), an FDA-approved drug for the treatment of multiple sclerosis, is a cytokine with immunomodulatory properties. Previous studies, including ours, demonstrated that IFNβ or type I IFN receptor signaling conferred protection against ischemic stroke in preclinical models, suggesting IFNβ might have translational therapeutic potential for the treatment of ischemic stroke. Currently, whether IFNβ could be coadministered with tPA to alleviate delayed tPA-induced adverse effects remains unknown. To elucidate that, IFNβ was coadministered with delayed tPA to ischemic stroke animals, and the severity and pathology of ischemic brain injury were assessed. We found delayed tPA treatment exacerbated ischemic brain injury, manifested by aggravated BBB disruption and HT. Notably, IFNβ ameliorated delayed tPA–exacerbated brain injury and alleviated adverse effects. Mechanistic studies revealed IFNβ suppressed tPA-enhanced neuroinflammation and MMP3/9 production in the ischemic brain. Furthermore, we identified IFNβ suppressed MMP9 production in microglia and attenuated tight junction protein degradation in brain endothelial cells. Moreover, we observed that peripheral immune cells may participate to a lesser extent in delayed tPA–exacerbated brain injury during the early phase of ischemic stroke. In conclusion, we provide the first evidence that IFNβ can be coadministered with tPA to mitigate delayed tPA–induced adverse effects of BBB disruption and HT that could potentially extend the tPA therapeutic window for the treatment of ischemic stroke.
Introduction
Stroke is a leading cause of death and results in permanent disability in up to 30% of survivors. There are 2 types of stroke: ischemic stroke accounts for ∼80% of cases, and hemorrhagic stroke accounts for 20%. Tissue plasminogen activator (tPA) is the only US Food and Drug Administration (FDA)–approved drug for ischemic stroke. Although tPA restores cerebral blood flow by dissolving blood clots in the ischemic brain, reperfusion that occurs after tPA treatment recruits peripheral inflammatory immune cells into the infarct core, leading to secondary brain injury.1 Furthermore, tPA was reported to induce microglia (MG) activation, which further exacerbates ischemic brain injury.2,3 Moreover, tPA administration beyond its therapeutic window of 3 to 4.5 hours postinjury induces serious adverse effects, resulting in increased risk of blood-brain barrier (BBB) disruption and hemorrhagic transformation (HT) in ischemic stroke.4-6
Interferon-β (IFNβ), a cytokine with immunomodulatory properties, was approved by the FDA for the treatment of multiple sclerosis (MS) more than a decade ago, and its anti-inflammatory properties are well characterized, suggesting that IFNβ has therapeutic potential for the treatment of ischemic stroke.7,8 Indeed, our previous study revealed that IFNβ attenuated ischemic brain injury in mice subjected to 40-minute middle cerebral artery occlusion (MCAO) and demonstrated that IFNβ modulated MG activation, diminished the central nervous system (CNS) infiltration of peripheral immune cells, inhibited adhesion molecule expression, and suppressed chemokines and MMP9 production in ischemic stroke.9 In addition, studies from other groups showed that IFNβ and/or type I IFN receptor (IFNAR1) signaling conferred protection against ischemic stroke in several animal models.10-15 Importantly, a recent study showed that increased IFNβ messenger RNA expression in the peripheral blood of stroke patients was strongly linked to a good outcome of stroke recovery.16 Altogether, these findings strongly demonstrate the beneficial effects of IFNβ for the treatment of ischemic stroke.
Currently, whether IFNβ could be coadministered with tPA to alleviate delayed tPA–induced adverse effects remains unknown. To elucidate this, we assessed the effect of IFNβ on delayed tPA–exacerbated ischemic brain injury in mice subjected to MCAO. Our results showed that IFNβ ameliorated ischemic brain injury and reduced mortality in delayed tPA–treated MCAO mice. Additional mechanistic studies revealed that IFNβ alleviated delayed tPA–aggravated BBB disruption and HT and that this alleviation might be mediated through its inhibitory effects on MMP3 and MMP9 production in the ischemic brain. Thus, our current study provides the first evidence that IFNβ exerts a beneficial effect on the alleviation of delayed tPA–induced adverse effects in ischemic stroke, suggesting that IFNβ can be coadministered with tPA to potentially extend the tPA therapeutic window for the treatment of ischemic stroke.
Materials and methods
Mice
All animal experimental procedures in this study were approved by the Purdue Animal Care and Use Committee and performed in strict compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. C57BL/6 mice used in this study were purchased from the Jackson Laboratory (Bar Harbor, ME). All mice were housed and bred with controlled humidity, temperature, and a 12-hour:12-hour light-dark cycle in the animal facility. Food and water were available ad libitum.
Reagents
IFNβ was purchased from PBL Interferon Source (Piscataway, NJ). tPA was purchased from Genentech (San Francisco, CA). Evans blue, Drabkin’s reagent, and hemoglobin were purchased from Sigma-Aldrich (St. Louis, MO). Triphenyltetrazolium chloride (TTC) and trichloroacetic acid were purchased from Alfa Aesar (Tewksbury, MA). Anti-MMP9 (clone L51/82) and anti-MMP3 (clone M4405F10) antibodies for western blots and Alexa Fluor 488 anti-CD45 (clone 30-F11), APC anti-CD11b (clone M1/70), phycoerythrin (PE)/Cy7 anti-Ly6G (clone 1A8), PE anti-CD3 (clone 145-2C11), PE anti-CD80 (clone 16-10A1), and PE/Cy7 anti-CD86 (clone GL-1) antibodies for flow cytometry were purchased from BioLegend (San Diego, CA). Alexa Fluor 488 anti-Iba1 antibody (ab225260) was purchased from Abcam (Cambridge, MA). Anti-occludin (13409-1-AP) and anti-MMP9 (10375-2-AP) antibodies for immunofluorescence staining were purchased from Proteintech (Chicago, IL). Anti-ZO-1 (61-7300), Alexa Fluor 488 anti-rabbit immunoglobulin G (IgG), and Alexa Fluor 594 anti-rabbit IgG antibodies for immunofluorescence staining were purchased from Invitrogen (Carlsbad, CA).
MCAO model
Cerebral ischemia was induced in adult male mice (age 3-4 months) and aged male and female mice (age 18-24 months) as previously described.9 Briefly, mice were subjected to intraluminal occlusion of the right middle cerebral artery by silicone-coated nylon monofilament (Doccol Corp, Sharon, MA). Cerebral blood flow was measured by laser Doppler flowmetry. The filament was withdrawn after 40-minute or 3-hour occlusion to allow reperfusion. Mouse body temperature was maintained at ∼37°C with a warming lamp and heating pad throughout the surgery. After surgery, mice were placed in a recovery cage where the temperature was maintained at 36°C for 1 hour. Mice with cerebral blood flow reduction >80% were included in the study and assigned randomly to receive IV administration of vehicle (phosphate-buffered saline [PBS]), tPA (5 mg/kg), or tPA plus IFNβ (10 000 U). The doses of tPA and IFNβ used in this study were based on previous studies.9,17,18 Our results showed that IFNβ did not affect the thrombolytic effect of tPA (supplemental Figure 1). The investigators who performed the assays were blinded to the experimental groups.
Infarct volume measurements
Mice were perfused with PBS, and the ischemic brain was harvested followed by 2-mm coronal slicing with a rodent brain matrix. After staining with 1% TTC, the brain sections were scanned and the infarct volume was calculated by using ImageJ as previously described.9
Evans blue extravasation assay
Mice were IV administered 4 mL/kg 2% Evans blue dye/0.9% saline solution (weight/volume) through the lateral tail vein. After 1 hour of circulation, mice were anesthetized and perfused with PBS to remove intravascular Evans blue. The brain was then harvested, sliced, and scanned. The hemispheres of the brain were separated and homogenized with 50% trichloroacetic acid solution. After centrifugation, the supernatant was collected and diluted with 95% ethanol at a ratio of 1:3. The amount of extravascular Evans blue in the supernatant was then calculated by measuring the fluorescence with excitation at 540/25 nm and emission at 645/40 nm (BioTek Synergy HT microplate reader).
Assessment of HT
HT was quantified with a spectrophotometric assay as previously described.19 After euthanasia, mice were perfused with PBS, and the ipsilateral hemisphere of the ischemic brain was harvested; 250 µL of distilled water was added to the brain tissue, followed by homogenization. After centrifugation, 20 µL of supernatant was mixed with 80 µL of Drabkin’s reagent to react for 15 minutes at room temperature. The supernatant was then subjected to the measurement of absorbance at a 540-nm wavelength (BioTek Synergy HT microplate reader).
Cell cultures
Primary MG were generated from neonatal mice as previous described.20 Briefly, cerebral cortical cells were collected from 1- to 2-day-old neonatal mice and seeded in T75 flasks containing DMEM/F12 complete medium. After removing medium, the flasks were replenished with complete medium containing 10 ng/mL of granulocyte-macrophage colony-stimulating factor on days 3 and 6 after plating. MG were harvested by shaking the flasks at 250 rpm for 30 minutes at 37°C on day 13 or 14. Mouse brain endothelial (bEnd.3) cells obtained from American Type Culture Collection were grown to confluence. After trypsinization, cells were seeded onto coverslips coated with poly-d-lysine followed by stimulation.
Mononuclear cell isolation and FACS analysis
Mice subjected to 3-hour MCAO or 40-minute MCAO were anesthetized and transcardially perfused with PBS at indicated time points. After removing the meninges, olfactory bulb, and cerebellum, the forebrains were homogenized with 1X HBSS buffer followed by filtration through a 70-μm nylon cell strainer. After centrifugation, cells were resuspended in 30% Percoll and underlayered with 70% Percoll. After centrifugation, the mononuclear cells were then isolated from the interface between 30% and 70% Percoll. Mononuclear cells isolated from 3-hour MCAO mice were stained with antibodies of CD45, CD11b, CD80, and CD86 to determine MG activation or CD45, CD11b, Ly6G, and CD3 to evaluate cell infiltrates by fluorescence-activated cell sorting (FACS) analysis. Mononuclear cells isolated from 40-minute MCAO mice were stained with antibodies of CD45, CD11b, and Ly6G followed by FACS analysis of cell infiltration (positive control).
Immunohistochemistry and histology
Mice were perfused with ice-cold PBS, and brain samples were dissected and fixed with 4% paraformaldehyde in PBS at 4°C overnight. After sucrose dehydration, brain samples were embedded in optimal cutting temperature and cut into 40-μm cryosections. After permeabilization, brain sections were stained with primary anti-MMP9 antibody (1:100) overnight at 4°C followed by secondary Alexa Flour 594 anti-rabbit IgG antibody at room temperature for 2 hours. After wash, brain sections were stained with Alexa Fluor 488 anti-Iba1 antibody (1:100) for 2 hours at room temperature followed by coverslipping with ProLong Gold antifade mountant containing 4′,6-diamidino-2-phenylindole (DAPI). The Z-stack images were visualized by confocal microscopy (Fluoview FV10i; Olympus). Paraffin-embedded brain tissues were sectioned and mounted on microscope slides. The slides were subjected to antigen retrieval, blocking, and staining with Alexa Fluor 488 anti-Iba1 antibody (1:100) for 2 hours. After wash, the slides were coverslipped with ProLong Gold antifade mountant containing DAPI. For histology, the slides were subjected to hematoxylin and eosin staining. Immunofluorescence and histology images were captured with fluorescence microscope (BX53; Olympus; camera: EXi Aqua; Q Imaging). The immunofluorescence images were then subjected to Iba1+ cell analysis by using ImageJ software.
Immunocytochemistry
bEnd.3 cells seeded on coverslips were fixed with 2% paraformaldehyde for 15 minutes at room temperature followed by permeabilization. After blocking, cells were then incubated with anti-Occludin (1:50) or anti-ZO-1 (1:100) for 2 hours at room temperature. After wash, cells were incubated with Alexa Fluor 488 goat anti-rabbit IgG for 1 hour at room temperature followed by coverslipping with ProLong Gold antifade mountant containing DAPI. Confocal images were acquired by using the Fluoview FV10i confocal microscope (Olympus). Quantification of fluorescent intensity was performed by using ImageJ software.
Quantitative polymerase chain reaction
Messenger RNA expression was measured by quantitative polymerase chain reaction analysis as previously described.21 The primers used were tumor necrosis factor α (TNFα): 5′-ATGGCCTCCCTCTCATCAGT-3′ and 5′-CTTGGTGGTTTGCTACGACG-3′; interleukin 1α (IL-1α): 5′-CGCTTGAGTCGGCAAAGAAAT-3′ and 5′-CTTCCCGTTGCTTGACGTTG-3′; IL-1β: 5′-CCCTGCAGCTGGAGAGTGTGGA-3′ and 5′-TGTGCTCTGCTTGGAGGTGCTG-3′; Ccl3: 5′-CCCAGCCAGGTGTCATTTTC-3′ and 5′-CTCAAGCCCCTGCTCTACAC-3′; Mmp9: 5′-AAAACCTCCAACCTCACGGA-3′ and 5′-GCGGTACAAGTATGCCTCTGC-3′; and Mmp3: 5′-TTCTGGGCTATACGAGGGCA-3′ and 5′-TTCTTCACGGTTGCAGGGAG-3′.
Western blot analysis
Brain tissue lysates were prepared in RIPA buffer with 0.3% sodium dodecyl sulfate (SDS). Proteins were separated on 10% SDS polyacrylamide gel electrophoresis. After transferring to polyvinylidene difluoride membranes, the blot was reacted with specific antibodies and detected by using Immobilon Western Chemiluminescent HRP Substrate (Millipore, Temecula, CA) as previously described.20
Gelatin zymography
MMP9 activity was measured by gelatin zymography as described.20 Briefly, the supernatants harvested from cell cultures were mixed with equal volumes of sample buffer and then analyzed by 10% SDS polyacrylamide gel electrophoresis containing 0.1% gelatin. After electrophoresis, the gel was washed in renaturing solution followed by incubation in developing buffer overnight at 37°C. The gel was then stained with staining solution and destained until clear bands were observed on the gel. Gelatinolytic activity was measured by quantifying the intensity of bands using imageJ.
Statistical analysis
All results in this study are given as means ± standard errors of the mean. Sample sizes were determined by power calculations based on our previous studies and preliminary results (power, 90%; α = 0.05). The normal distribution of the data was confirmed by Shapiro-Wilk test. Comparisons between 2 groups were performed by the unpaired Student t test, whereas comparisons among multiple groups were performed by 1-way analysis of variance followed by the Bonferroni post hoc test. For the data that are not normally distributed, comparisons between 2 or multiple groups were calculated with the use of the Mann-Whitney U or Kruskal-Wallis test followed by Dunn’s post hoc test, respectively. Statistical significance was determined with P < .05.
Results
Delayed but not early tPA treatment exacerbates ischemic brain injury
To determine whether delayed tPA treatment affects brain injury in ischemic stroke, mice were subjected to 40-minute MCAO followed by tPA administration at 40 minutes postinjury (early treatment) or 3 hours postreperfusion (delayed treatment). At 24 hours postinjury, the ischemic brains were harvested and subjected to TTC staining to determine the infarct volume. Our results showed that tPA administered at 40 minutes postinjury did not affect the outcome of ischemic brain injury, because the infarct volume of tPA-treated MCAO mice was similar to that of vehicle-treated MCAO controls (Figure 1A). In contrast, tPA administered at 3 hours postreperfusion exacerbated ischemic brain injury in 40-minute MCAO mice. The infarct volume of tPA-treated MCAO mice was increased more than twofold compared with that of vehicle-treated MCAO controls (Figure 1B). Notably, IFNβ coadministered with tPA at 3 hours postreperfusion ameliorated delayed tPA–exacerbated brain injury. The infarct volume of tPA plus IFNβ-treated MCAO mice was significantly decreased compared with that of tPA-treated MCAO mice (Figure 1B). Collectively, these results demonstrate that delayed tPA treatment exacerbates ischemic brain injury, whereas IFNβ exerts a protection against delayed tPA–exacerbated brain injury in ischemic stroke.
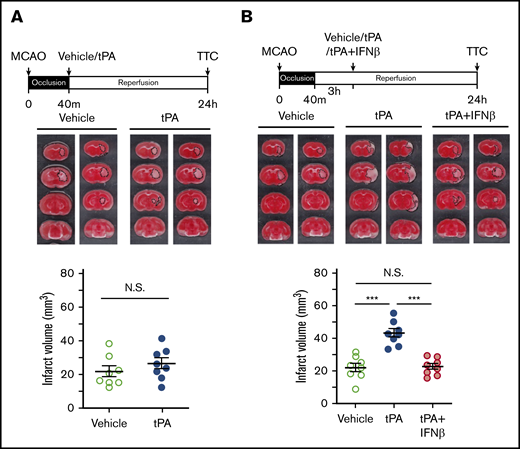
Delayed but not early tPA treatment exacerbates ischemic brain injury. (A) Mice were subjected to 40-minute occlusion and administered vehicle or tPA at 40 minutes postinjury (n = 8 per group). (B) Mice subjected to 40-minute occlusion were administered vehicle or tPA in the presence or absence of IFNβ at 3 hours postreperfusion (n = 8 per group). 24 hours after disease onset, ischemic brains were harvested and subjected to TTC staining. Two representative TTC-stained brain samples of each group are shown. The infarct volume of ischemic brains was also measured. ***P < .001 by 1-way analysis of variance (ANOVA). Not significant (N.S.) by unpaired Student t test (A) or 1-way ANOVA (B).
Delayed but not early tPA treatment exacerbates ischemic brain injury. (A) Mice were subjected to 40-minute occlusion and administered vehicle or tPA at 40 minutes postinjury (n = 8 per group). (B) Mice subjected to 40-minute occlusion were administered vehicle or tPA in the presence or absence of IFNβ at 3 hours postreperfusion (n = 8 per group). 24 hours after disease onset, ischemic brains were harvested and subjected to TTC staining. Two representative TTC-stained brain samples of each group are shown. The infarct volume of ischemic brains was also measured. ***P < .001 by 1-way analysis of variance (ANOVA). Not significant (N.S.) by unpaired Student t test (A) or 1-way ANOVA (B).
IFNβ ameliorates delayed tPA–exacerbated brain injury and reduces mortality in ischemic stroke
To closely mimic the clinical condition, mice were subjected to 3-hour MCAO followed immediately by vehicle, IFNβ, tPA, or tPA plus IFNβ administration. At 20 hours postinjury, the ischemic brains were harvested to assess infarct volume. Our results showed that mice subjected to 3-hour MCAO exhibited severe brain injury with increased infarct volume compared with mice subjected to 40-minute MCAO (Figures 1B and 2A). We then evaluated the effect of IFNβ on ischemic brain injury in 3-hour MCAO mice. Our results showed that IFNβ did not offer protection against ischemic brain injury in 3-hour MCAO mice at day 1 postinjury, because IFNβ-treated MCAO mice displayed a similar infarct volume as vehicle-treated MCAO controls at 20 hours poststroke (supplemental Figure 2A). However, the protective effect of IFNβ in ischemic stroke was observed at day 2 postinjury. We found that the infarct volume of IFNβ-treated MCAO mice was significantly lower than that of vehicle-treated MCAO controls at 45 hours postinjury (supplemental Figure 2A). The protective effect of IFNβ on the attenuation of infarct volume in 3-hour MCAO mice may be attributed to its effect on the suppression of peripheral immune cell infiltration, because the number of CD45hiCD11b+ infiltrating peripheral immune cells in the ischemic brain of IFNβ-treated MCAO mice was significantly lower than that of vehicle-treated MCAO controls (supplemental Figure 2B).
![IFNβ ameliorates delayed tPA-exacerbated brain injury and reduces mortality in ischemic stroke. (A) Adult male mice were subjected to 3-hour MCAO followed immediately by vehicle, tPA, or tPA plus IFNβ administration. At 20 hours postinjury, the ischemic brains were harvested and then subjected to TTC staining. Two representative TTC-stained brain samples of each group are shown, and the infarct volume of ischemic brains was measured (n = 10 per group). The survival rates of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice were monitored up to day 7 postischemia (n = 10 per group). Aged male (B) and aged female (C) mice were subjected to 3-hour MCAO followed by vehicle, tPA, or tPA plus IFNβ administration. Mice were euthanized at 20 hours postinjury, and the harvested ischemic brains were subjected to TTC staining. Two representative TTC-stained brain samples of each group are shown, and the infarct volume of ischemic brains was measured (aged male mice: vehicle, n = 7; tPA, n = 8; tPA plus IFNβ, n = 10; aged female mice: vehicle, n = 9; tPA, n = 7; tPA plus IFNβ, n = 7). The mortality rate was also assessed (the numbers indicated on the figures represent the number of MCAO mice that died/the number of MCAO mice included in each group). *P < .05 by 1-way ANOVA (A [left],C) or log-rank test of Kaplan-Meier survival curve (A, right), **P < .01 by 1-way ANOVA (A [left]-B).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/18/10.1182_bloodadvances.2020001443/3/m_advancesadv2020001443f2.png?Expires=1768034593&Signature=GyCFO0OGzPMMYyQUHS-lKARfDPs4cx8YIWSHClegabL3dG9c0b-gU~IY1go6zWY2tbkRvCQ3B882-3zEwTGNI6mhBALD6BGm11~6qpFSxAydJLpGODsJHOnvmxKYi3Vvttl74w2W~hhfw8sS27~MmCANfZgpCwGsEbUpyZfTm1JC70-rsDIefKG86xEln2qo~duhl3sh8f84u~GbFQwHP6XiqXzEth0ONOj74aJZrF8YZhW1f1eUwwiivByjjUStps04hg4VVec1myUmVMi2yFLIRHu7azj3MWJqdo6rhQuRBbXFE7oOb~gxU~~KlehaWKFAacsxs0boClVHmpqepw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
IFNβ ameliorates delayed tPA-exacerbated brain injury and reduces mortality in ischemic stroke. (A) Adult male mice were subjected to 3-hour MCAO followed immediately by vehicle, tPA, or tPA plus IFNβ administration. At 20 hours postinjury, the ischemic brains were harvested and then subjected to TTC staining. Two representative TTC-stained brain samples of each group are shown, and the infarct volume of ischemic brains was measured (n = 10 per group). The survival rates of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice were monitored up to day 7 postischemia (n = 10 per group). Aged male (B) and aged female (C) mice were subjected to 3-hour MCAO followed by vehicle, tPA, or tPA plus IFNβ administration. Mice were euthanized at 20 hours postinjury, and the harvested ischemic brains were subjected to TTC staining. Two representative TTC-stained brain samples of each group are shown, and the infarct volume of ischemic brains was measured (aged male mice: vehicle, n = 7; tPA, n = 8; tPA plus IFNβ, n = 10; aged female mice: vehicle, n = 9; tPA, n = 7; tPA plus IFNβ, n = 7). The mortality rate was also assessed (the numbers indicated on the figures represent the number of MCAO mice that died/the number of MCAO mice included in each group). *P < .05 by 1-way ANOVA (A [left],C) or log-rank test of Kaplan-Meier survival curve (A, right), **P < .01 by 1-way ANOVA (A [left]-B).
IFNβ ameliorates delayed tPA-exacerbated brain injury and reduces mortality in ischemic stroke. (A) Adult male mice were subjected to 3-hour MCAO followed immediately by vehicle, tPA, or tPA plus IFNβ administration. At 20 hours postinjury, the ischemic brains were harvested and then subjected to TTC staining. Two representative TTC-stained brain samples of each group are shown, and the infarct volume of ischemic brains was measured (n = 10 per group). The survival rates of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice were monitored up to day 7 postischemia (n = 10 per group). Aged male (B) and aged female (C) mice were subjected to 3-hour MCAO followed by vehicle, tPA, or tPA plus IFNβ administration. Mice were euthanized at 20 hours postinjury, and the harvested ischemic brains were subjected to TTC staining. Two representative TTC-stained brain samples of each group are shown, and the infarct volume of ischemic brains was measured (aged male mice: vehicle, n = 7; tPA, n = 8; tPA plus IFNβ, n = 10; aged female mice: vehicle, n = 9; tPA, n = 7; tPA plus IFNβ, n = 7). The mortality rate was also assessed (the numbers indicated on the figures represent the number of MCAO mice that died/the number of MCAO mice included in each group). *P < .05 by 1-way ANOVA (A [left],C) or log-rank test of Kaplan-Meier survival curve (A, right), **P < .01 by 1-way ANOVA (A [left]-B).
We then analyzed the effect of delayed tPA treatment in 3-hour MCAO mice. Consistently, we found tPA treatment at 3 hours postinjury exacerbated brain injury with increased infarct volume compared with vehicle treatment in 3-hour MCAO mice at 20 hours postinjury (Figure 2A). Notably, the coadministration of IFNβ with tPA at 3 hours postinjury was able to ameliorate delayed tPA–exacerbated ischemic brain injury in 3-hour MCAO mice. The infarct volume of tPA plus IFNβ-treated MCAO mice was significantly decreased compared with that of tPA-treated MCAO mice at day 1 postinjury (Figure 2A). In addition, the long-term survival rate of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice was assessed. Our results showed that vehicle- and tPA plus IFNβ-treated MCAO mice exhibited similar survival rates; however, tPA-treated MCAO mice displayed a decreased survival rate compared with vehicle- and tPA plus IFNβ-treated MCAO mice. At day 7 postinjury, tPA-treated MCAO mice had a survival rate of only 10%, whereas vehicle- and tPA plus IFNβ-treated MCAO mice had a survival rate of 40% (Figure 2A).
Both age and sex have been reported to affect the outcome of brain injury in ischemic stroke.22 To assess whether IFNβ exerts protection against delayed tPA–exacerbated brain injury in aged animals of both sexes, aged male and female mice were subjected to 3-hour MCAO followed by vehicle, tPA, or tPA plus IFNβ administration, and the infarct volume and mortality rate were then assessed. Similarly, delayed tPA treatment further exacerbated brain injury with increased infarct volume and mortality rate in both aged male and female stroke animals. Consistently, IFNβ exerted protection against delayed tPA–exacerbated brain injury, because it reduced infarct volume and mortality rate in both delayed tPA–treated aged male and female MCAO mice (Figure 2B-C). Altogether, these results demonstrate that IFNβ can be coadministered with tPA to ameliorate delayed tPA–exacerbated brain injury in adult male stroke animals and aged stroke animals of both sexes.
IFNβ alleviates neuroinflammation in delayed tPA–treated ischemic stroke
We previously showed that IFNβ modulated neuroinflammation in ischemic stroke through inhibiting MG activation and suppressing inflammatory cytokine production.9 To evaluate whether IFNβ modulates neuroinflammation in delayed tPA–treated ischemic stroke, mice were subjected to 3-hour MCAO followed by administration of vehicle, tPA, or tPA plus IFNβ, and the ischemic brains were then harvested and subjected to FACS analysis and immunohistochemistry to assess MG activation and quantitative polymerase chain reaction to determine inflammatory mediator production. Mononuclear cells isolated from the ischemic brain of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice were subjected to FACS analysis to assess the expression of surface maturation markers CD80 and CD86 on MG. MG were determined based on their intermediate expression of CD45 and positive expression of CD11b. Our results showed tPA increased CD80+ and CD86+ MG, whereas IFNβ repressed tPA-increased CD80+ and CD86+ MG in the ischemic brain of 3-hour MCAO mice (Figure 3A). We then assessed the number of Iba1+ cells in the ipsilateral cortex and striatum of stroke animals; we observed the number of Iba1+ cells in the ipsilateral cortex and striatum of ischemic brain was increased in the tPA-treated MCAO mice compared with vehicle-treated MCAO controls. Notably, the number of Iba1+ cells in the ipsilateral cortex and striatum of tPA plus IFNβ-treated MCAO mice was significantly lower than that in tPA-treated MCAO mice (Figure 3B), indicating IFNβ suppressed MG activation in delayed tPA–treated ischemic stroke. Finally, inflammatory cytokine production in the ischemic brain was measured. Our results showed that tPA enhanced the expression of inflammatory cytokines TNFα, IL-1α, and IL-1β, chemokine CCL3, and metalloproteinases MMP3 and MMP9 in the ischemic brain. In contrast, IFNβ suppressed tPA-induced upregulation of the aforementioned inflammatory mediators in the ischemic brain (Figure 3C). Altogether, our results demonstrate that delayed tPA treatment enhances MG activation and neuroinflammation in ischemic stroke. In contrast, IFNβ mitigates those adverse effects when coadministered with tPA.
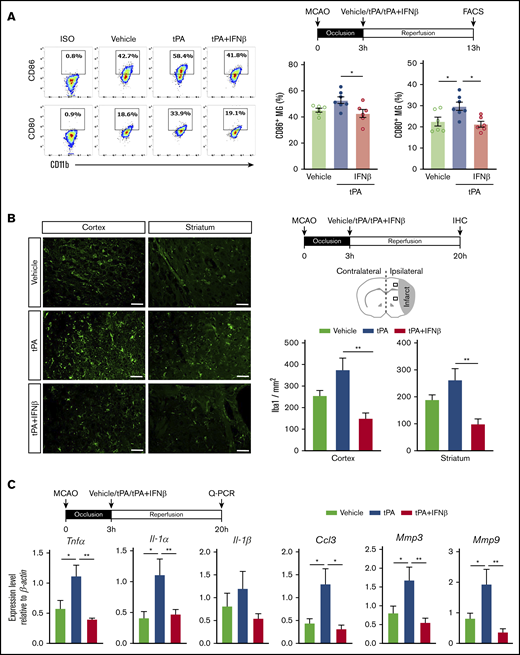
IFNβ alleviates neuroinflammation in delayed tPA–treated ischemic stroke. Mice were subjected to 3-hour MCAO followed by IV administration of vehicle, tPA, or tPA plus IFNβ. (A) At 13 hours postinjury, mononuclear cells were isolated from the ischemic brain of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice (n = 6-7 per group), and the isolated cells were then stained with antibodies of CD45, CD11b, CD80, and CD86 followed by FACS analysis. MG were determined based on their expression of CD45intCD11b+, and MG with positive expression of CD80 and CD86 were then determined. Isotype controls (ISO) were used as a negative control to determine positive expression of CD80 or CD86. (B) At 20 hours postinjury, the brain tissues harvested from vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice were subjected to immunohistochemistry (IHC), and Iba1+ cells in the ipsilateral cortex and striatum of periinfarct regions were quantified (n = 6 per group). Scale bars, 50 µm. (C) The brain tissues were also subjected to quantitative polymerase chain reaction (Q-PCR) analysis for IL-1α, IL-1β, TNFα, CCL3, MMP3, and MMP9 messenger RNA expression (n = 5-8 per group). *P < .05 by 1-way ANOVA, **P < .01 by Kruskal-Wallis test (B) or 1-way ANOVA (C).
IFNβ alleviates neuroinflammation in delayed tPA–treated ischemic stroke. Mice were subjected to 3-hour MCAO followed by IV administration of vehicle, tPA, or tPA plus IFNβ. (A) At 13 hours postinjury, mononuclear cells were isolated from the ischemic brain of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice (n = 6-7 per group), and the isolated cells were then stained with antibodies of CD45, CD11b, CD80, and CD86 followed by FACS analysis. MG were determined based on their expression of CD45intCD11b+, and MG with positive expression of CD80 and CD86 were then determined. Isotype controls (ISO) were used as a negative control to determine positive expression of CD80 or CD86. (B) At 20 hours postinjury, the brain tissues harvested from vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice were subjected to immunohistochemistry (IHC), and Iba1+ cells in the ipsilateral cortex and striatum of periinfarct regions were quantified (n = 6 per group). Scale bars, 50 µm. (C) The brain tissues were also subjected to quantitative polymerase chain reaction (Q-PCR) analysis for IL-1α, IL-1β, TNFα, CCL3, MMP3, and MMP9 messenger RNA expression (n = 5-8 per group). *P < .05 by 1-way ANOVA, **P < .01 by Kruskal-Wallis test (B) or 1-way ANOVA (C).
IFNβ alleviates delayed tPA–aggravated BBB disruption and HT in ischemic stroke
tPA administration beyond its therapeutic window has been shown to aggravate BBB disruption and induce HT in ischemic stroke.4,23 We investigated whether IFNβ amelioration of delayed tPA–exacerbated ischemic brain injury is mediated through alleviation of delayed tPA–aggravated BBB disruption and HT in ischemic stroke. We first evaluated the effect of IFNβ on delayed tPA–aggravated BBB disruption. Mice were subjected to 3-hour MCAO followed by vehicle, tPA, or tPA plus IFNβ administration, and the Evans blue leakage of ischemic brain was assessed. Our results showed that 3-hour MCAO induced BBB disruption, resulting in Evans blue leakage into the ipsilateral but not contralateral hemisphere of ischemic brain (Figure 4A). tPA treatment exaggerated Evans blue leakage in the ischemic brain (Figure 4A). Notably, IFNβ was able to abolish tPA-aggravated BBB disruption, because the level of Evans blue leakage in the ischemic brain of tPA plus IFNβ-treated stroke animals was similar to that in vehicle-treated stroke controls (Figure 4A).
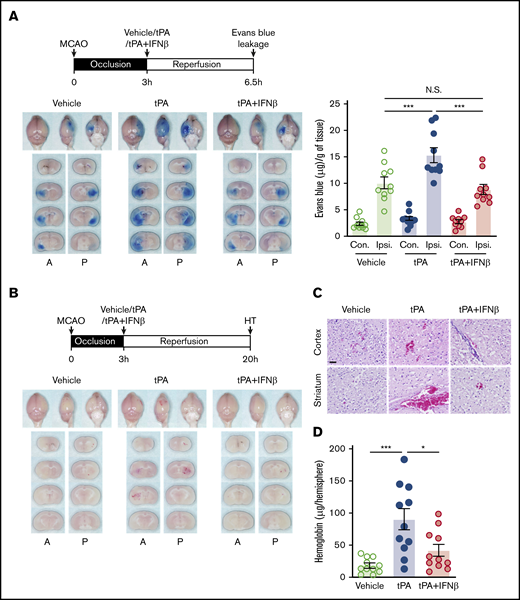
IFNβ alleviates delayed tPA–aggravated BBB disruption and HT in ischemic stroke. (A) Mice were subjected to 3-hour MCAO followed by IV administration of vehicle, tPA, or tPA plus IFNβ. Mice were then injected with Evans blue 1 hour before euthanization, and the ischemic brains were harvested at 6.5 hours postinjury followed by imaging and sectioning. The Evans blue leakage in the contralateral (Con) and ipsilateral (Ipsi) hemispheres was quantified (n = 10 per group). (B) Three-hour MCAO mice were administered with vehicle, tPA, or tPA plus IFNβ at 3 hours postinjury. The ischemic brains were harvested at 20 hours postinjury followed by sectioning and imaging. The representative brain section images are shown. (C) The representative hematoxylin and eosin staining images of 3 independent experiments are shown. Scale bar, 50 µm. (D) The levels of hemoglobin in the ipsilateral hemisphere of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice were measured (n = 11 per group). *P < .05, ***P < .001 by 1-way ANOVA. A, anterior surface; P, posterior surface.
IFNβ alleviates delayed tPA–aggravated BBB disruption and HT in ischemic stroke. (A) Mice were subjected to 3-hour MCAO followed by IV administration of vehicle, tPA, or tPA plus IFNβ. Mice were then injected with Evans blue 1 hour before euthanization, and the ischemic brains were harvested at 6.5 hours postinjury followed by imaging and sectioning. The Evans blue leakage in the contralateral (Con) and ipsilateral (Ipsi) hemispheres was quantified (n = 10 per group). (B) Three-hour MCAO mice were administered with vehicle, tPA, or tPA plus IFNβ at 3 hours postinjury. The ischemic brains were harvested at 20 hours postinjury followed by sectioning and imaging. The representative brain section images are shown. (C) The representative hematoxylin and eosin staining images of 3 independent experiments are shown. Scale bar, 50 µm. (D) The levels of hemoglobin in the ipsilateral hemisphere of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice were measured (n = 11 per group). *P < .05, ***P < .001 by 1-way ANOVA. A, anterior surface; P, posterior surface.
We then assessed whether IFNβ exerts protection against delayed tPA–induced HT in ischemic stroke. Mice were subjected to 3-hour MCAO followed by vehicle, tPA, or tPA plus IFNβ administration, and the level of HT in the ischemic brain was evaluated. We observed that tPA treatment induced HT in the ischemic brain (Figure 4B). In addition, hematoxylin and eosin staining showed a large number of red blood cells extravasated into the cortex and striatum of tPA-treated MCAO mice (Figure 4C). Finally, we confirmed that the hemoglobin level in the ipsilateral hemisphere of tPA-treated MACO mice was significantly higher than that in vehicle-treated MCAO controls (Figure 4D). In contrast, IFNβ mitigated tPA-induced HT, leading to a considerable reduction of red blood cell extravasation and hemoglobin level in the ischemic brain (Figure 4B-D). Altogether, these results demonstrate that delayed tPA treatment aggravates BBB disruption and induces HT. In contrast, IFNβ alleviates the aforementioned adverse effects induced by delayed tPA treatment in ischemic stroke.
IFNβ suppresses tPA-enhanced MMP3 and MMP9 expression in the ischemic brain
A previous study showed that MMP9 played a detrimental role in BBB disruption.24 In addition, MMP3 has been shown to exert effects on the induction of HT in tPA-treated ischemic stroke and exacerbation of HT in hyperglycemic stroke.25,26 Therefore, we assessed whether IFNβ modulates MMP3 and MMP9 production in delayed tPA–treated stoke animals. Because cerebral ischemia has been shown to induce biphasic BBB disruption,27,28 we therefore measured MMP3 and MMP9 expression at the early (6 and 9 hours postinjury) and late (20 hours postinjury) time points after ischemic stroke. During the early time point of ischemic stroke, a low level of MMP3 and MMP9 expression was observed in the ipsilateral but not contralateral hemisphere of vehicle-treated stroke animals (Figure 5A). tPA treatment further enhanced MMP3 and MMP9 expression in the ipsilateral hemisphere of stroke animals, and tPA had a greater effect on upregulating the expression of MMP3 than MMP9. In contrast, IFNβ suppressed tPA-enhanced MMP3 and MMP9 expression in the ischemic brain (Figure 5B; supplemental Figure 3). During the late phase of ischemic stroke, similar results were observed in which tPA enhanced MMP3 and MMP9 expression, and IFNβ suppressed tPA-enhanced MMP3 and MMP9 expression in the ischemic brain (Figure 5C). Altogether, our results demonstrate that IFNβ suppresses delayed tPA–enhanced MMP3 and MMP9 expression that may contribute to its effects on the alleviation of BBB disruption and HT in delayed tPA–treated stroke animals.
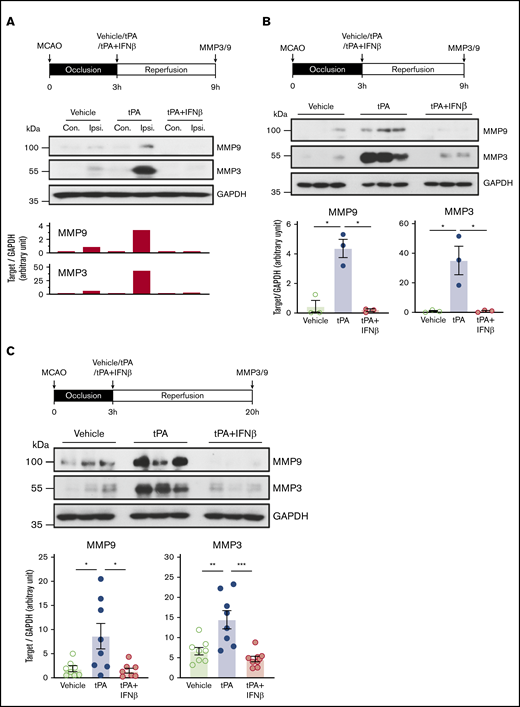
IFNβ suppresses tPA-enhanced MMP3 and MMP9 expression in the ischemic brain. Mice were subjected to 3-hour MCAO followed by vehicle, tPA, or tPA plus IFNβ administration. (A) At 9 hours postinjury, the ischemic brains were harvested and separated into the contralateral and ipsilateral hemispheres followed by western blot analysis. The representative results of MMP9 and MMP3 expression in the contralateral (Con) and ipsilateral (Ipsi) hemispheres of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice are shown. (B) At 9 hours postinjury, MMP3 and MMP9 expression in the ipsilateral hemisphere of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice (n = 3 per group) was measured by western blots. Statistical analysis was based on the comparison between vehicle and tPA or tPA and tPA plus IFNβ. (C) At 20 hours postinjury, the ipsilateral hemisphere of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice was harvested and subjected to western blot analysis for MMP3 and MMP9 expression. The representative results of MMP9 and MMP3 expression in the ipsilateral hemisphere of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice are shown (n = 3 per group), and the level of MMP9 and MMP3 expression was also quantified (n = 8 per group). *P < .05 by Kruskal-Wallis test (B) or 1-way ANOVA (C), **P < .01, ***P < .001 by 1-way ANOVA. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
IFNβ suppresses tPA-enhanced MMP3 and MMP9 expression in the ischemic brain. Mice were subjected to 3-hour MCAO followed by vehicle, tPA, or tPA plus IFNβ administration. (A) At 9 hours postinjury, the ischemic brains were harvested and separated into the contralateral and ipsilateral hemispheres followed by western blot analysis. The representative results of MMP9 and MMP3 expression in the contralateral (Con) and ipsilateral (Ipsi) hemispheres of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice are shown. (B) At 9 hours postinjury, MMP3 and MMP9 expression in the ipsilateral hemisphere of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice (n = 3 per group) was measured by western blots. Statistical analysis was based on the comparison between vehicle and tPA or tPA and tPA plus IFNβ. (C) At 20 hours postinjury, the ipsilateral hemisphere of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice was harvested and subjected to western blot analysis for MMP3 and MMP9 expression. The representative results of MMP9 and MMP3 expression in the ipsilateral hemisphere of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice are shown (n = 3 per group), and the level of MMP9 and MMP3 expression was also quantified (n = 8 per group). *P < .05 by Kruskal-Wallis test (B) or 1-way ANOVA (C), **P < .01, ***P < .001 by 1-way ANOVA. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
IFNβ inhibits MMP9 production in MG and attenuates TJP degradation in brain endothelial cells
Because MG were reported to secrete MMP9 after ischemic stroke,29 we evaluated whether tPA enhances MMP9 expression in MG and whether IFNβ suppresses tPA-enhanced MMP9 expression in MG in the ischemic brain. Our results showed tPA enhanced MMP9 expression and increased MMP9-expressing Iba1+ cells in the ipsilateral cortex and striatum of MCAO mice. In contrast, IFNβ suppressed tPA-enhanced MMP9 expression and repressed MMP9-expressing Iba1+ cells in the ischemic brain (Figure 6A). To further confirm our in vivo findings, we evaluated the effect of tPA and IFNβ on MMP9 production in primary MG. Because TNFα and PGE2 are potent MMP9 inducers and can be detected in the ischemic brain,21,30-32 we then treated MG with TNFα plus PGE2 or TNFα plus PGE2 plus tPA in the presence or absence of IFNβ followed by western blot analysis and gelatin zymography to determine MMP9 production and activity, respectively. Our results showed that TNFα plus PGE2 induced MMP9 production and activity in MG, and tPA further enhanced MMP9 production and activity in TNFα plus PGE2–treated MG. In contrast, IFNβ abolished tPA-enhanced MMP9 production and activity in MG (Figure 6B-C). In addition, to determine whether IFNβ possesses a protective effect on the attenuation of tight junction protein (TJP) degradation, bEnd.3 cells stimulated with TNFα or TNFα plus tPA in the presence or absence of IFNβ were subjected to immunohistochemical analysis to assess the expression levels of TJP, including ZO-1 and occludin. TNFα and TNFα plus tPA induced substantial degradation of ZO-1 and occludin expression in bEnd.3 cells. In contrast, IFNβ attenuated TNFα plus tPA–induced degradation of ZO-1 and occludin expression in bEnd.3 cells (Figure 6D-E). Collectively, our results demonstrate that IFNβ suppresses MMP9 expression in MG and attenuates TJP degradation in brain endothelial cells.
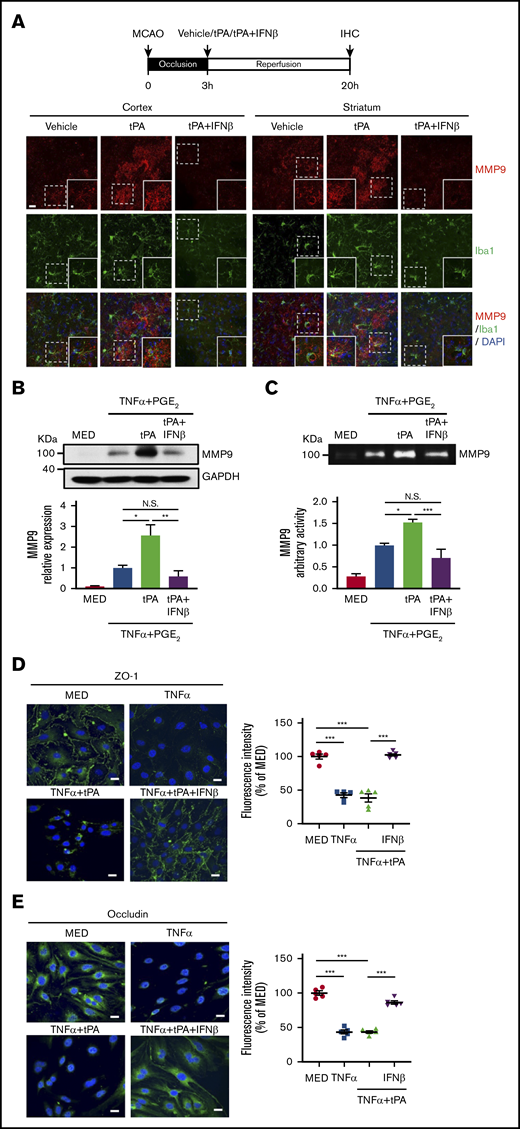
IFNβ inhibits MMP9 production in MG and attenuates TJP degradation in brain endothelial cells. (A) Mice were subjected to 3-hour MCAO followed by vehicle, tPA, or tPA plus IFNβ administration. At 20 hours postinjury, the ischemic brains were harvested and subjected to IHC analysis for MMP9 and Iba1 expression (n = 5 per group). The representative confocal images of the cortex and striatum of ipsilateral hemisphere of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice are shown. Scale bar, 20μm (5 μm in magnified boxes). (B) Primary MG were treated with 20 ng/mL of TNFα plus 10−6 M PGE2 or TNFα plus PGE2 plus tPA (10 µg/mL) in the presence or absence of IFNβ (1000 U/mL) for 24 hours. Cells were then collected and subjected to western blots to measure MMP9 expression (n = 5 per group). (C) Supernatants were collected and subjected to gelatin zymography to determine MMP9 activity (n = 5 per group). (D-E) bEnd.3 cells were treated with TNFα (50 ng/mL in panel D and 100 ng/mL in panel E) or TNFα plus tPA (10 µg/mL) in the presence or absence of IFNβ (1000 U/mL). 24 hours later, cells were subjected to immunocytochemistry for ZO-1 and occludin expression. The representative confocal images are shown, and the fluorescence intensity of ZO-1 and occludin was also quantified (n = 5). Scale bars, 20 μm. *P < .05, **P < .01, ***P < .001 by 1-way ANOVA. N.S., no significant difference by 1-way ANOVA.
IFNβ inhibits MMP9 production in MG and attenuates TJP degradation in brain endothelial cells. (A) Mice were subjected to 3-hour MCAO followed by vehicle, tPA, or tPA plus IFNβ administration. At 20 hours postinjury, the ischemic brains were harvested and subjected to IHC analysis for MMP9 and Iba1 expression (n = 5 per group). The representative confocal images of the cortex and striatum of ipsilateral hemisphere of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice are shown. Scale bar, 20μm (5 μm in magnified boxes). (B) Primary MG were treated with 20 ng/mL of TNFα plus 10−6 M PGE2 or TNFα plus PGE2 plus tPA (10 µg/mL) in the presence or absence of IFNβ (1000 U/mL) for 24 hours. Cells were then collected and subjected to western blots to measure MMP9 expression (n = 5 per group). (C) Supernatants were collected and subjected to gelatin zymography to determine MMP9 activity (n = 5 per group). (D-E) bEnd.3 cells were treated with TNFα (50 ng/mL in panel D and 100 ng/mL in panel E) or TNFα plus tPA (10 µg/mL) in the presence or absence of IFNβ (1000 U/mL). 24 hours later, cells were subjected to immunocytochemistry for ZO-1 and occludin expression. The representative confocal images are shown, and the fluorescence intensity of ZO-1 and occludin was also quantified (n = 5). Scale bars, 20 μm. *P < .05, **P < .01, ***P < .001 by 1-way ANOVA. N.S., no significant difference by 1-way ANOVA.
Peripheral immune cells may participate to a lesser extent in delayed tPA–exacerbated brain injury during the early phase of ischemic stroke
Cerebral reperfusion after ischemia promotes the CNS infiltration of peripheral immune cells, including neutrophils, monocytes/macrophages, and T cells, which subsequently leads to secondary brain injury. To elucidate whether infiltrating immune cells play a role in delayed tPA–exacerbated ischemic brain injury, mice were subjected to 3-hour MCAO followed by administration with vehicle, tPA, or tPA plus IFNβ. At 13 hours postinjury, mononuclear cells were isolated from the ischemic brain of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice and then subjected to FACS analysis to determine the total infiltrating myeloid immune cells (CD45hiCD11b+), neutrophils (CD45hiCD11b+Ly6G+), and T cells (CD45hiCD11b−CD3+; Figure 7A). Our results showed that a low number of CD45hiCD11b+ cells were detected in the ischemic brain of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice compared with positive control MCAO mice (Figure 7B). In addition, unlike positive control MCAO mice, which exhibited a large number of CD45hiCD11b+ cells in the ipsilateral hemisphere compared with contralateral hemisphere, we did not observe a significant difference in the number of CD45hiCD11b+ cells between the ipsilateral and contralateral hemispheres among vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice (Figure 7B). Further analysis revealed that a few neutrophils and T cells were detected in the ischemic brain of vehicle-, tPA-, and tPA plus IFNβ-treated MCAO mice, and there was no significant difference among them (Figure 7C-D). Taken together, our results demonstrate that tPA treatment does not promote the recruitment of the CNS-infiltrating immune cells, suggesting peripheral immune cells may participate to a lesser extent in delayed tPA–exacerbated ischemic brain injury during the early phase of the disease.
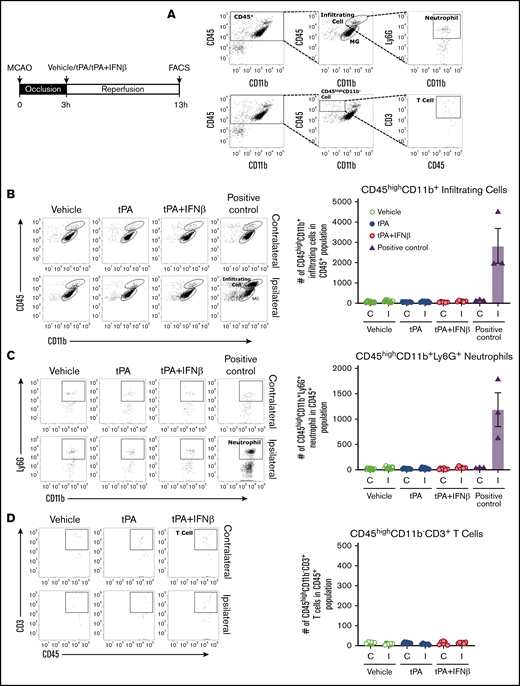
Peripheral immune cells may participate to a lesser extent in delayed tPA–exacerbated brain injury during the early phase of ischemic stroke. Mice were subjected to 3-hour MCAO followed by vehicle, tPA, or tPA plus IFNβ administration (n = 6-7 per group) or subjected to 40-minute MCAO as positive control (n = 3). Three-hour MCAO mice administered vehicle, tPA, or tPA plus IFNβ were euthanized at 13 hours postinjury, and 40-minute MCAO mice were euthanized at 48 hours postinjury. The ischemic brains were harvested and subjected to mononuclear cells isolation. The isolated mononuclear cells were then subjected to staining with antibodies against CD45, CD11b, Ly6G, and CD3 followed by FACS analysis. (A) The infiltrating myeloid immune cells were determined based on their expression of CD45hiCD11b+, and neutrophils were determined based on their positive expression of Ly6G within the population of CD45hiCD11b+ cells. T cells were determined based on their positive expression of CD3 within the population of CD45hiCD11b− cells. The numbers of total infiltrating myeloid immune cells (B), neutrophils (C), and T cells (D) in the contralateral and ipsilateral hemispheres of ischemic brains were analyzed. C, contralateral hemisphere; I, ipsilateral hemisphere.
Peripheral immune cells may participate to a lesser extent in delayed tPA–exacerbated brain injury during the early phase of ischemic stroke. Mice were subjected to 3-hour MCAO followed by vehicle, tPA, or tPA plus IFNβ administration (n = 6-7 per group) or subjected to 40-minute MCAO as positive control (n = 3). Three-hour MCAO mice administered vehicle, tPA, or tPA plus IFNβ were euthanized at 13 hours postinjury, and 40-minute MCAO mice were euthanized at 48 hours postinjury. The ischemic brains were harvested and subjected to mononuclear cells isolation. The isolated mononuclear cells were then subjected to staining with antibodies against CD45, CD11b, Ly6G, and CD3 followed by FACS analysis. (A) The infiltrating myeloid immune cells were determined based on their expression of CD45hiCD11b+, and neutrophils were determined based on their positive expression of Ly6G within the population of CD45hiCD11b+ cells. T cells were determined based on their positive expression of CD3 within the population of CD45hiCD11b− cells. The numbers of total infiltrating myeloid immune cells (B), neutrophils (C), and T cells (D) in the contralateral and ipsilateral hemispheres of ischemic brains were analyzed. C, contralateral hemisphere; I, ipsilateral hemisphere.
Discussion
tPA, the only FDA-approved drug for acute ischemic stroke, exerts its therapeutic effect by dissolving blood clots in the ischemic brain. However, tPA has a limited therapeutic window and has to be administered within 3 to 4.5 hours from the onset of stroke to be effective.33 Studies indicate that <10% of patients with acute ischemic stroke are able to receive tPA treatment because of its limited therapeutic window.34,35 Delayed treatment with tPA has been shown to exacerbate BBB disruption and induce HT, leading to enhanced ischemic brain injury.36 In addition, tPA was reported to aggravate neuroinflammation through activating MG in ischemic stroke.2,3 Therefore, developing a therapy that can be coadministered with tPA to ameliorate neuroinflammation and extend the tPA therapeutic window is urgent and necessary.
We previously reported that IFNβ, an FDA-approved treatment for MS, ameliorated brain injury and lessened neuroinflammation in animals subjected to 40-minute MCAO.9 In the present study, we investigated whether IFNβ can be coadministered with tPA to ameliorate delayed tPA–exacerbated brain injury in ischemic stroke. We first evaluated the effect of early (40 minutes postinjury) and delayed (3 hours postreperfusion) tPA treatment on ischemic brain injury using MCAO models with 40-minute occlusion. We found that early and delayed tPA treatment resulted in different outcomes of brain injury in MCAO animals. Early treatment with tPA did not alter the outcome of ischemic brain injury. In contrast, delayed tPA treatment exacerbated ischemic brain injury, with significantly increased infarct volume compared with vehicle treatment. Our findings are consistent with previous studies37,38 and demonstrate that delayed tPA treatment exacerbates ischemic brain injury.
Although we observed that delayed tPA treatment exacerbated brain injury in MCAO mice subjected to 40-minute occlusion, tPA was administered to 40-minute MCAO mice at 3 hours postreperfusion. To closely mimic the clinical condition, we then conducted MCAO models with 3-hour occlusion followed immediately by tPA treatment and assessed the ischemic brain injury. Under this condition, we also observed that tPA exacerbated ischemic brain injury. Therefore, these results further confirmed the detrimental effect of delayed tPA treatment in ischemic stroke and demonstrated the feasibility of using 3-hour MCAO models followed by tPA treatment to mimic ischemic brain injury exacerbated by delayed tPA treatement in ischemic stroke.
Using 3-hour MCAO animal models allowed us to test the therapeutic efficacy of IFNβ on modulating delayed tPA–exacerbated ischemic brain injury. We observed IFNβ, coadministered with tPA, ameliorated delayed tPA–exacerbated brain injury and attenuated delayed tPA–enlarged infarct volume to a level similar to that in vehicle-treated stroke controls, demonstrating the protective effect of IFNβ on delayed tPA–exacerbated ischemic brain injury. Because both age and sex have been reported to affect the outcome of ischemic stroke,22 we also evaluated the effect of IFNβ on delayed tPA treatment in both aged male and female MCAO mice. Aged male MCAO mice exhibited enhanced brain injury compared with adult male and aged female MCAO mice. Delayed tPA treatment exacerbated brain injury in both aged male and female MCAO mice. Importantly, the protective effect of IFNβ on the amelioration of ischemic brain injury could still be observed in both aged male and female MCAO mice with delayed tPA treatment. Thus, our findings strongly demonstrate that IFNβ can be coadministered with tPA to ameliorate delayed tPA–exacerbated ischemic brain injury, and the protective effect of IFNβ on delayed tPA–treated ischemic stroke is not limited by age or sex.
Previously, we showed that IFNβ attenuated ischemic brain infarct in 40-minute MCAO mice at day 2 postinjury.9 In this study, we observed IFNβ lessened ischemic brain injury in 3-hour MCAO mice at day 2 but not day 1 postinjury. Interestingly, we found that IFNβ was able to ameliorate delayed tPA–exacerbated ischemic brain injury at day 1 postinjury. Thus, these results suggest that IFNβ-offered protection against delayed tPA–treated ischemic stroke may be largely due to its direct effects on the alleviation of tPA-induced adverse effects that subsequently lessen tPA-exacerbated ischemic brain injury. Because we did not observe that IFNβ ameliorated ischemic brain injury in 3-hour MCAO mice at day 1 postinjury, we speculated that 2 major factors may be involved. Firstly, prolonged cerebra occlusion induced severe brain injury in 3-hour MCAO animals. The infarct volume of 3-hour MCAO mice (Figure 2A) increased more than twofold compared with that of 40-minute MCAO mice at day 1 poststroke (Figure 1A). Thus, this level of severity in brain injury may have abolished the protective effect of IFNβ in ischemic stroke. Secondly, one of the protective mechanisms of IFNβ in 40-minute MCAO mice was to suppress the peripheral immune cells infiltrating into the ischemic brain.9 However, we did not observe a significant increase of peripheral immune cells in the ischemic brain of 3-hour MCAO mice at day 1 postinjury (Figure 7), indicating that the severity of brain injury observed in 3-hour MCAO mice was largely due to prolonged occlusion, and peripheral immune cells might participate to a lesser extent in promoting ischemic brain injury during the early phase of the disease under this condition. Indeed, previous studies reported that peripheral immune cells were largely recruited into the ischemic brain at day 2 but not day 1 poststroke.39-41 Similarly, we observed a considerable number of peripheral immune cells infiltrating into the ischemic brain of 3-hour MCAO mice at day 2 but not day 1 postinjury (supplemental Figures 2B and 7B). Consistent with our previous findings,9 we observed IFNβ suppressed peripheral immune cell infiltration and attenuated cerebral infarct in 3-hour MCAO mice at day 2 postinjury (supplemental Figure 2). Altogether, these results further confirm the protective effect of IFNβ in ischemic stroke.
Delayed tPA treatment has been linked to exaggerated BBB disruption and induction of HT in ischemic stroke, and MMP9 and MMP3 were reported to be the culprits of inducing those adverse effects.24,25,42 Similarly, we also observed delayed tPA treatment aggravated BBB disruption, induced HT, and enhanced MMP3 and MMP9 expression in the ischemic brain. Importantly, when IFNβ was coadministered with tPA to ischemic stroke animals, IFNβ was able to alleviate delayed tPA–aggravated BBB disruption and HT in the ischemic brain. Because previous studies reported that IFNβ suppressed MMP9 production in MS and ischemic stroke,9,43,44 it prompted us to investigate whether IFNβ-mediated alleviation of delayed tPA–induced adverse effects is mediated through modulating MMP3 and MMP9 production in the ischemic brain. Indeed, our results demonstrated that IFNβ inhibited both MMP9 and MMP3 production in the ischemic brain of delayed tPA–treated stroke animals, and that further revealed a novel function of IFNβ on the suppression of MMP3 production. Altogether, these results suggest that IFNβ-mediated suppression of MMP3 and MMP9 production may contribute to its protective effects on lessening delayed tPA–exaggerated BBB disruption and HT in ischemic stroke.
Previous studies showed that the CNS-resident cells, including MG, astrocytes, and brain endothelial cells, produced MMP3 and/or MMP9 after tPA treatment in ischemic stroke.45-47 In addition, infiltrating peripheral immune cells, especially neutrophils, were reported to produce MMP9 in the ischemic brain.48 In this study, we observed that tPA enhanced MMP3 and MMP9 expression in the ischemic brain as early as 6 hours postinjury (supplemental Figure 3). However, we did not observe increased cell infiltrates in the ischemic brain of tPA-treated MCAO mice compared with vehicle-treated MCAO controls at 13 hours postinjury (Figure 7), suggesting that the CNS-resident cells may be largely responsible for the increased MMP3 and MMP9 production in the ischemic brain of tPA-treated stroke animals, although the contribution of MMP3 and MMP9 production from infiltrating immune cells could not be fully excluded. tPA has been shown to bind to LDL receptor related protein-1 that subsequently activates the PDGF-CC/PDGFRα→NFκB/ERK pathway, leading to the upregulation of MMP3 and MMP9 production.47,49 Because our in vivo results demonstrated that IFNβ inhibited tPA-enhanced MMP3 and MMP9 expression in the ischemic brain, additional studies would be required to elucidate whether IFNβ regulates the tPA-activated LDL receptor related protein-1→PDGF-CC/PDGFRα→NFκB/ERK pathway to suppress MMP3 and MMP9 production in the ischemic brain after ischemic insults.
Nevertheless, there are limitations to our current study that may be worth considering. First, the intraluminal suture MCAO model was used in this study to evaluate the effect of IFNβ on the amelioration of delayed tPA–exacerbated ischemic brain injury. However, using the suture MCAO model limited us to assessing the thrombolytic effect of tPA in vivo. Therefore, future studies using the thromboembolic MCAO model would be necessary to confirm our findings of the protective effect of IFNβ on delayed tPA–exacerbated ischemic brain injury, because this would be clinically relevant to assess the beneficial effect of IFNβ on the extension of the tPA therapeutic window. Second, because we observed that IFNβ suppressed tPA-enhanced MMP3 and MMP9 expression and repressed MMP9+ Iba1+ cells in the ischemic brain of delayed tPA–treated MCAO mice, future studies using either IFNAR1 knockout or conditional IFNAR1 knockout in MG (CX3CR1CreERT2:IFNAR1fl/fl) are warranted to further confirm the suppressive effect of IFNβ on tPA-induced MMP3 and MMP9 production in the ischemic brain. Third, previous studies established the critical role of MMP3 and MMP9 in tPA-mediated HT and edema in mice subjected to photothrombotic MCAO and in hypertensive rats subjected to intraluminal suture MCAO, respectively.24,25 However, direct evidence demonstrating that MMP3 and MMP9 play an essential role in tPA-exacerbated HT and BBB disruption in normal animals subjected to intraluminal suture MCAO is still lacking. Therefore, future studies using MMP3- and MMP9-deficient mice to confirm their essential roles in tPA-induced adverse effects in animals subjected to intraluminal suture MCAO would be beneficial, because that would allow us to further confirm whether IFNβ-mediated suppression of MMP3 and MMP9 accounts for the major effects on the alleviation of tPA-induced adverse effects in ischemic stroke. Finally, it is possible that IFNβ-offered protection against delayed tPA–treated ischemic stroke is mediated through the combined effects of modulating MG activation, neuroinflammation, MMP3/9 production, and TJP degradation in the ischemic brain. Comprehensive mechanistic studies would be required to further dissect each specific effect of IFNβ on the alleviation of tPA-exacerbated ischemic brain injury.
In summary, we report for the first time that IFNβ, an FDA-approved drug for MS treatment, can be coadministered with tPA to ameliorate delayed tPA–exacerbated ischemic brain injury. We show IFNβ suppresses MG activation, modulates neuroinflammation, and inhibits MMP3/9 production that may subsequently lead to lessened BBB disruption and HT and attenuated brain injury in delayed tPA–treated ischemic stroke. Thus, our findings reveal a novel function of IFNβ on the alleviation of delayed tPA–induced adverse effects in ischemic stroke and open a new venue for the potential use of IFNβ combined with tPA to extend the tPA therapeutic window for ischemic stroke treatment.
For data sharing requests, e-mail the corresponding author, Jui-Hung Yen (jimyen@iu.edu).
Acknowledgment
This study was supported by research funding from grant R01NS102449 from National Institute of Neurological Disorders and Stroke, National Institutes of Health (J.-H.Y.).
Authorship
Contribution: P.-C.K. performed the experiments, analyzed data, and wrote the manuscript; W.-T.W. performed the experiments and analyzed data; B.A.S., D.F., H.C.P., A.J.I., and K.D.B. performed the experiments; I.-C.Y. designed the experiments and edited the manuscript; J.-H.Y. conceived the study, designed the experiments, and wrote the manuscript; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jui-Hung Yen, Department of Microbiology and Immunology, Indiana University School of Medicine, 2101 E Coliseum Blvd, Fort Wayne, IN 46805; e-mail: jimyen@iu.edu.

