

Key Points
A novel algorithm, RHtyper, provides comprehensive and high-throughput Rh blood group genotyping from whole-genome sequencing data.

Abstract
RHD and RHCE genes encode Rh blood group antigens and exhibit extensive single-nucleotide polymorphisms and chromosome structural changes in patients with sickle cell disease (SCD). RH variation can drive loss of antigen epitopes or expression of new epitopes, predisposing patients with SCD to Rh alloimmunization. Serologic antigen typing is limited to common Rh antigens, necessitating a genetic approach to detect variant antigen expression. We developed a novel algorithm termed RHtyper for RH genotyping from existing whole-genome sequencing (WGS) data. RHtyper determined RH genotypes in an average of 3.4 and 3.3 minutes per sample for RHD and RHCE, respectively. In a validation cohort consisting of 57 patients with SCD, RHtyper achieved 100% accuracy for RHD and 98.2% accuracy for RHCE, when compared with genotypes obtained by RH BeadChip and targeted molecular assays and after verification by Sanger sequencing and independent next-generation sequencing assays. RHtyper was next applied to WGS data from an additional 827 patients with SCD. In the total cohort of 884 patients, RHtyper identified 38 RHD and 28 RHCE distinct alleles, including a novel RHD DAU allele, RHD* 602G, 733C, 744T 1136T. RHtyper provides comprehensive and high-throughput RH genotyping from WGS data, facilitating deconvolution of the extensive RH genetic variation among patients with SCD. We have implemented RHtyper as a cloud-based public access application in DNAnexus (https://platform.dnanexus.com/app/RHtyper), enabling clinicians and researchers to perform RH genotyping with next-generation sequencing data.
Introduction
Alloimmunization to non-ABO antigens is one of the most common complications of red blood cell (RBC) transfusions.1 To prevent alloimmunization in patients with sickle cell disease (SCD), in addition to ABO and RhD, most centers transfuse Rh (C, E or C/c, E/e)- and K-antigen–matched RBCs. A minority of transfusion services also match for Fya/Fyb, Jka/Jkb, and S antigens. Serologic matching for Rh antigens reduces but does not eliminate Rh alloimmunization. Diversity of the RH gene among patients and blood donors of African descent is a risk factor for Rh alloimmunization and is not addressed by current serologic antigen-matching strategies.2,3
The Rh blood group system comprises 2 genes, RHD and RHCE, which encode 2 multipass transmembrane proteins carrying D, C; and/or c, E; and/or e antigens.4 However, the Rh system is more complex than these 5 common antigens. RHD and RHCE are duplicated genes with 97% sequence identity and exhibit extensive single-nucleotide polymorphisms (SNPs) and genetic rearrangements, resulting in expression of variant Rh antigens. These are associated with decreased expression of proteins (weak antigens), loss of common epitopes (partial antigens), and expression of new epitopes.5 Patients with variant Rh antigens are at risk of forming antibodies against Rh epitopes that are absent on their RBCs. More than 50 Rh-variant antigens have been described serologically, and more than 500 RHD and 100 RHCE alleles have been identified (https://www.isbtweb.org/; accessed 30 March 2018 for all data reported herein) with the number of newly identified alleles continuing to grow.6 RH variation is prevalent in individuals of African descent.2,7,8 A study of >1500 patients with SCD and African American blood donors revealed that 29% of RHD and 53% of RHCE alleles differ from those common in Europeans.2 A study of 500 Brazilian patients with SCD and African Brazilian donors reported that altered RHCE alleles are inherited with altered RHD alleles in 15% of patients and 7.8% of donors,7 and similar findings were described in France.8 Serologic antigen typing does not distinguish all RH alleles, and thus, preventing Rh alloimmunization potentially necessitates some level of genotype matching.
RH genotyping is currently performed by a variety of laboratory-developed tests such as SNP-based assays.9,10 These assays target the most prevalent alleles but are not comprehensive. DNA sequencing-based approaches, including whole-genome sequencing (WGS) and whole-exome sequencing (WES), offer advantages over the SNP-based platforms, including the capacity to identify new and rare SNPs, insertions/deletions (indels), and genetic rearrangements.11-15 However, published genotyping algorithms for analysis of next-generation sequencing (NGS) data involve multiple computational tools or software and require computationally intensive and laborious manual curation, rendering them difficult for application to large-scale data.12,15,16 To address the computational challenges for high-throughput RH genotyping, we developed an algorithm, RHtyper, for rapid and comprehensive RH genotyping from existing WGS data.
Methods
Patients
Existing WGS data from 884 patients treated at St Jude Children’s Research Hospital (St Jude) and Texas Children’s Hospital were used in this study. The St Jude patients had been enrolled in the Sickle Cell Clinical Research and Intervention Program (SCCRIP) study (www.clinicaltrials.gov NCT02098863).17 SCCRIP is a lifetime longitudinal cohort study, in which clinical information is prospectively collected and biologic samples are banked, including blood for genomics and proteomics studies. The present study was approved by the institutional review boards of St Jude and Baylor College of Medicine, and all participants or guardians provided written informed consent.
WGS and RH SNP array genotyping
Genomic DNA was extracted from peripheral blood mononuclear cells by standard methods, and WGS was performed at HudsonAlpha Institute for Biotechnology, as previously described.18,19 Paired-end reads were aligned against the human genome build GRCh38 (hg38) with the Burrows-Wheeler Aligner software package.20 The average sequencing coverage of WGS was 35.7×. RHD and RHCE genotyping with RH BeadChip and targeted molecular assays were performed, as previously described,2 for 57 of the 884 patients for RHtyper validation.
Development of RHtyper
The RHtyper algorithm was developed as shown in Figure 1A. An RH allele database was curated from the International Society of Blood Transfusion (ISBT) database (https://www.isbtweb.org, last accessed 30 March 2018 for all data reported herein) and the now-retired NCBI Blood Group Antigen Gene Mutation (BGMUT) database.21-23 The 2 databases do not include silent nucleotide variations with occasional exceptions. Our consolidated database included 415 RHD and 130 RHCE alleles annotated for genotype determination. The alleles from the ISBT and BGMUT databases are specified according to conventional RH mRNA sequences (RHD, L08429, and RHCE, DQ322275). Of note, the conventional RH mRNA sequences, which reflect known population variation,13 differ from the reference genomic sequence (hg38) by 2 SNPs in the coding region (conventional RHD mRNA, c.1136T, reference genomic sequence, c.1136C, and conventional RHCE mRNA, c. 48G, reference genomic sequence, c. 48C). Identified variants were first assigned on the basis of the reference genomic sequence and then modified to match the conventional sequences.
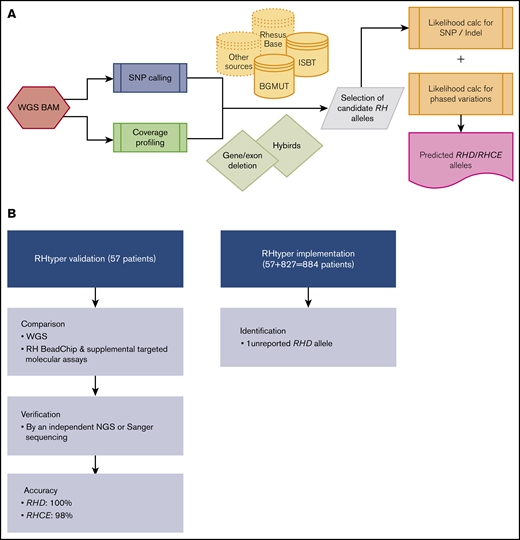
RHtyper development, validation, and implementation. (A) Overview of the RHtyper algorithm. (B) Validation (n = 57 patients) and implementation (n = 884 patients) of the RHtyper. BAM, binary alignment map.
RHtyper development, validation, and implementation. (A) Overview of the RHtyper algorithm. (B) Validation (n = 57 patients) and implementation (n = 884 patients) of the RHtyper. BAM, binary alignment map.
The RHtyper algorithm consists of variant calling; determination of RHD zygosity, allele zygosity, and hybrid alleles by coverage profiling; and prediction of RH allele pairs via likelihood scoring and reported haplotype associations (Figure 1A).
RH variant calling.
Variants were called via the SAMtools pileup method,24 using WGS reads that met predefined read criteria (base read quality, ≥15; mapping read quality, ≥10; and average read quality, ≥15). Counts of A, T, G, and C nucleotides and indels were generated for each exonic position of RHD/RHCE genes. The exonic positions with at least 7 reads harboring variant/alternative nucleotides (∼20% of variant fraction at 30× WGS coverage) were classified as confident heterozygous sites. SNPs and indels were annotated on the basis of encoded amino acid changes.
Determination of RHD zygosity and allele zygosity by coverage profiling.
For RHD zygosity, a sliding window coverage analysis with a bin size of 300 bp and step size of 150 bp was performed across the RH loci. The log2-transformed ratio of the median coverage per bin and genome-wide coverage was calculated. A bin was inferred as a homozygous deletion when the bin-based ratio was lower than −3 or as a heterozygous deletion when the ratio was lower than −0.6. If the number of homozygous deletion bins was larger than 60% of the total number of bins for RHD, homozygous deletion RHD was inferred. Similarly, if the number of heterozygous deletion bins was larger than 60% of the total number of bins for RHD, heterozygous deletion of RHD was inferred.
Allele zygosity, when 2 copies were present, was determined by the zygosity of SNPs. The fraction of the alternative nucleotide was determined by dividing the number of reads with the alternative nucleotide by the total number of reads at the indicated position. Heterozygous SNPs were called if the fraction of the alternative nucleotide was between 0.2 and 0.8, and homozygous SNPs were called if the fraction was >0.8. Heterozygous RH alleles were then inferred if heterozygous SNPs were present; otherwise, homozygous RH alleles were inferred. Because the exon 8 sequence of conventional RHD is identical with that of RHCE (c.1136C) and the reference genomic sequence is RHD*DAU0 (c.1136T), the sequence reads from conventional RHD exon 8 are more likely to align with RHCE than with the reference RHD, leading to low coverage of reference RHD exon 8 and increased coverage of RHCE exon 8. Therefore, for SNPs of exon 8 (eg, RHD c.1136 C>T), the fraction of the alternative nucleotide was determined by dividing the reads containing the alternative nucleotide by genome-wide average read depth and coverage, rather than the position-specific read depth and coverage.
Identification of RH hybrid allele boundaries and hybrid alleles assisted by coverage profiling.
The potential boundaries of RH hybrid alleles were determined by the circular binary segmentation (CBS) algorithm.25,26 Specifically, the algorithm segmented the bin-based coverage profile of a sample recursively, to assess the mean coverage difference between 2 consecutive segments bordering a change point. The change point was deemed significant if the value of the mean coverage difference between the 2 consecutive segments was P < .05, by paired Student t test. The boundaries of the segments were determined by identifying the largest segment surrounded by 2 significant change points. The copy number of each exon was then determined according to the log2-transformed ratio of the exonic medians between RHD and RHCE. Thresholds of −2, −0.6, 0.4, 0.8, and 1.2 were used to call 2-copy loss, 1-copy loss, no loss or gain, 1-copy gain, and 2-copy gain, respectively. Hybrid alleles were predicted by combining the exon copy number profiles, hybrid allele-characteristic SNPs, and allele-pair predictions.
Prediction of RH allele pairs by using likelihood scoring and reported haplotype associations.
Confirmation by Sanger sequencing
To confirm novel RH SNPs identified by RHtyper, the RHD and RHCE exons containing the SNPs were amplified by polymerase chain reaction (PCR) with gene-specific primers (supplemental Methods). The PCR products were then separated by electrophoresis, and the products with the expected lengths were purified and sent for Sanger sequencing.
Results
Validation of RHtyper
Using existing WGS data, we developed a Bayesian likelihood-based computational algorithm termed RHtyper for characterization of RH genotypes (Figure 1A). We first validated RHtyper with WGS data from a cohort of 57 patients with SCD whose RH genotypes had been determined by RH BeadChip and targeted molecular assays detecting common altered alleles, as described previously (Figure 1B).2 Sanger sequencing and independent NGS assays were performed on samples with discrepancies and on those with additional SNPs and indels found by RHtyper. In this validation cohort, RHtyper identified all the genetic changes tested and identified by the RH BeadChip and targeted molecular assays with the following exceptions (Table 1): (1) RHtyper identified 2 hemizygous RHD deletions that PCR-based zygosity testing failed to detect, which was confirmed by independent NGS assays; and (2) RHtyper discovered 1 additional RHCE c.48G>C SNP that was not confirmed by Sanger sequencing. Further investigation noted a SNP at the binding site of the 5′ PCR primer for Sanger sequencing that was potentially responsible for the absence of confirmation. Among genetic changes not tested by the RH BeadChip and targeted molecular assays, RHtyper identified 3 missense SNPs that were all confirmed by independent NGS assays: RHD c.473G>A (novel, n = 1), RHD c.520G>C (n = 1), and RHCE c.941T>C (n = 2; Table 1). Five distinct silent SNPs and indels (not shown) were also detected: RHD c.541C>T (novel; n = 1), RHD c.579 G>A (n = 3), RHD c.819G>A (n = 2), RHD c.329-330TG deletion (n = 1), and RHCE c.105C>T (n = 5). All the silent SNPs and indels were confirmed by independent NGS assays, except for 1 sample with RHCE c.105C>T. The missense SNP (RHD c.473G>A, p.Ser158Asn) and silent SNP (RHD c.541C>T/p.Leu181Leu) appear to be novel, in that they have not been reported in published RH genotype databases.
Protein encoding changes identified additionally by RHtyper analysis of WGS in a validation cohort of 57 patients with SCD
| Additional changes identified by RHtyper | RHD | RHCE | ||||||
|---|---|---|---|---|---|---|---|---|
| Nucleotide | Amino acid | Discordant, n | Confirmed, n* | Nucleotide | Amino acid | Discordant, n. | Confirmed, n* | |
| Tested by targeted RH SNP genotyping† | ||||||||
| Zygosity | Hemizygous deletion | — | 2 | 2 | — | — | — | — |
| Missense | — | — | — | — | 48 G>C | Trp16Cys | 1 | 0 |
| Not tested by targeted RH SNP genotyping | ||||||||
| Missense | 473 G>A (novel) | Ser158Asn | 1 | 1 | 941 T>C | Val314Ala | 2 | 2 |
| 520 G>C | Val174Met | 1 | 1 | |||||
| Additional changes identified by RHtyper | RHD | RHCE | ||||||
|---|---|---|---|---|---|---|---|---|
| Nucleotide | Amino acid | Discordant, n | Confirmed, n* | Nucleotide | Amino acid | Discordant, n. | Confirmed, n* | |
| Tested by targeted RH SNP genotyping† | ||||||||
| Zygosity | Hemizygous deletion | — | 2 | 2 | — | — | — | — |
| Missense | — | — | — | — | 48 G>C | Trp16Cys | 1 | 0 |
| Not tested by targeted RH SNP genotyping | ||||||||
| Missense | 473 G>A (novel) | Ser158Asn | 1 | 1 | 941 T>C | Val314Ala | 2 | 2 |
| 520 G>C | Val174Met | 1 | 1 | |||||
Confirmation by Sanger sequencing or independent NGS assays.
RHD and RHCE BeadChip and targeted molecular assays.11
RHtyper next determined alleles based on the identified SNPs and indels, coverage profiling, and established haplotype associations and allele frequencies. Among the 57 patients, RHtyper identified 14 different RHD and 13 different RHCE alleles. Compared with alleles identified by the RH BeadChip and targeted molecular assays and after confirmation by additional Sanger sequencing and independent NGS assays performed for SNP/indel and RHD zygosity verification, the allele prediction accuracy of RHtyper was 100% (114 of 114 alleles) for RHD and 98.2% (112 of 114 alleles) for RHCE. The 2 differences in RHCE alleles were: (1) RHCE*ce by BeadChip and Sanger sequencing vs RHCE*ce48C by RHtyper; and (2) RHCE*ce48C by BeadChip and NGS vs RHCE*ce 48C, 105T (silent) by RHtyper. These differences were related to the 2 unconfirmed SNPs detected by RHtyper (RHCE c.48G>C, n = 1; RHCE c.105C>T, n = 1).
Prediction of RH genotypes of patients with RHtyper
We applied RHtyper to the WGS data from an additional 827 patients with SCD, providing a total cohort of 884 patients (Figure 1B). RHtyper determined the RH genotypes with an average run time per sample of 3.4 minutes (range, 0.1-5.2 minutes) for RHD and 3.3 minutes (range, 2.2-5.3 minutes) for RHCE. Overall, RHtyper identified 41 different RHD and 28 different RHCE SNPs and indels among the total cohort of 884 individuals (Figure 2). A second novel missense SNP (RHD c.364T>A, p.Ser122Thr) and 5 more novel silent SNPs were identified, all of which were confirmed by Sanger sequencing. In total, we identified 2 missense and 6 silent RH SNPs in the 884 patients that had not been reported (Table 2). Further sequencing read analysis on the 2 novel missense SNPs showed that RHD c.364T>A was linked to RHD*pseudogene and thus would not be expressed. RHD c.473G>A could not be phased but was also identified in an individual with RHD*pseudogene.
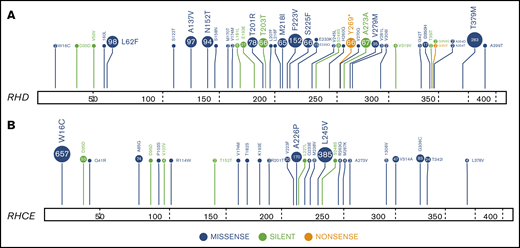
Landscape of RH gene SNPs and indels in 884 patients with SCD. RHtyper identified 41 different RHD (A) and 28 different RHCE (B) SNPs and indels.
Landscape of RH gene SNPs and indels in 884 patients with SCD. RHtyper identified 41 different RHD (A) and 28 different RHCE (B) SNPs and indels.
Novel RH gene SNPs identified in 884 patients with SCD
| Gene | Exon | Coding position | Class | Amino acid position | Samples, n |
|---|---|---|---|---|---|
| RHD | 1 | 105 C>T | Silent | Asp35Asp | 1 |
| 3 | 364 T>A* | Missense | Ser122Thr | 1 | |
| 3 | 473 G>A* | Missense | Ser158Asn | 1 | |
| 4 | 541 C>T | Silent | Leu181Leu | 6 | |
| RHCE | 2 | 285 C>T | Silent | Asp95Asp | 1 |
| 2 | 321 C>T | Silent | Val107Val | 4 | |
| 3 | 456 C>T | Silent | Thr152Thr | 2 | |
| 5 | 681 G>C | Silent | Leu227Leu | 1 |
| Gene | Exon | Coding position | Class | Amino acid position | Samples, n |
|---|---|---|---|---|---|
| RHD | 1 | 105 C>T | Silent | Asp35Asp | 1 |
| 3 | 364 T>A* | Missense | Ser122Thr | 1 | |
| 3 | 473 G>A* | Missense | Ser158Asn | 1 | |
| 4 | 541 C>T | Silent | Leu181Leu | 6 | |
| RHCE | 2 | 285 C>T | Silent | Asp95Asp | 1 |
| 2 | 321 C>T | Silent | Val107Val | 4 | |
| 3 | 456 C>T | Silent | Thr152Thr | 2 | |
| 5 | 681 G>C | Silent | Leu227Leu | 1 |
RHD pseudogene (RHD*Ψ) associated.
RHtyper next determined alleles based on the identified SNPs and indels, coverage profiling, and the established haplotype associations and allele frequencies. Among the 884 patients, RHtyper identified 38 distinct RHD alleles (Table 3). The 3 most frequently found RHD alleles were conventional RHD (n = 920; frequency, 0.5204), RHD deletion (n = 294; 0.1663), and RHD*DAU0, c.1136T (n = 229; 0.1295), a finding that is similar to previous reports of individuals with SCD and African American blood donors.2,3 RHtyper determined RHD zygosity by coverage profiling (Figure 3A). Using characteristic SNPs supplemented by coverage profiling, RHtyper identified 65 RHD*DIIIa-CEVS(4-7)-D hybrid alleles (n = 65; 0.0368), in which RHD*DIIIa exons 4 to 7 are replaced by RHCE exons 4 to 7.6 One RHD*D-CE(4–7)-D hybrid allele with no SNPs (n = 1; 0.006) was found based on coverage profiling only and was subsequently confirmed by quantitative PCR (Figure 3B; supplemental Table 1).30 In addition, RHtyper predicted hybrid allele boundaries by the CBS algorithm using coverage profiling. As proof of concept, we used the predicted boundaries as a reference, identified the actual breakpoints of RHD*DIIIa-CEVS(4-7)-D in 2 samples (SJSCD040920 and SJSCD04093; supplemental Figure 1) by Sanger sequencing.
RHD and RHCE Alleles identified in 884 patients with SCD
| Allele ISBT name | Allele detail/alias | Allele, n | Allele frequency |
|---|---|---|---|
| RHD alleles | |||
| RHD*01 | — | 920 | 0.5204 |
| RHD Deletion | — | 294 | 0.1663 |
| RHD*10.00 RHD*DAU0 | RHD*1136T | 229 | 0.1295 |
| RHD*08N.01RHD*Pseudo- gene RHD*Ψ | — | 66 | 0.0373 |
| RHD*03N.01 | RHD*DIIIa-CEVS (4-7)-D | 65 | 0.0368 |
| RHD*09.03.01 RHD*DAR3.01 | RHD*602G,667G | 57 | 0.0322 |
| RHD*10.03 RHD*DAU3 | RHD*835A,1136T | 35 | 0.0198 |
| RHD*10.00.01 RHD*DAU0.01 | RHD*1136T,579A | 21 | 0.0119 |
| RHD*04.01 RHD*DIVa | RHD*186T,410T, 455C,1048C | 16 | 0.0090 |
| RHD*10.05 RHD*DAU5 | RHD*667G,697C,1136T | 12 | 0.0068 |
| RHD*03.01 RHD*DIIIa | RHD*186T,410T,455C,602G,667G,819A | 10 | 0.0057 |
| No ISBT designation (proposed as RHD*03.01.02 RHD*DIII.01.02) | RHD*186T,410T,455C,602G,667G* | 7 | 0.0040 |
| RHD*01W.33RHD*weak D type 33 | RHD*520A | 3 | 0.0017 |
| RHD*09.01.02 RHD*DAR1.02 | RHD*602G,667G, 1025C | 3 | 0.0017 |
| No ISBT designation | RHD*841C (GenBank: KU363613.2)† | 3 | 0.0017 |
| RHD*01.01 | RHD*48C | 2 | 0.0011 |
| RHD*01W.66RHD*weak D type 66 | RHD*916A | 2 | 0.0011 |
| RHD*12.01 RHD*DOL1 | RHD*509C,667G | 2 | 0.0011 |
| RHD*49 RHD*DWN | RHD*1053T, 1057T, 1059G, 1060A, 1061A | 2 | 0.0011 |
| RHD* 01W.137RHD*weak D type 137 | RHD*780A | 1 | 0.0006 |
| RHD*01N.07 | RHD*D-CE (4-7)-D | 1 | 0.0006 |
| RHD*01N.35 | RHD*330_331delGT | 1 | 0.0006 |
| RHD*01W.1RHD*weak D type 1 | RHD*809G | 1 | 0.0006 |
| RHD*01W.45RHD*weak D type 45 | RHD*1195A | 1 | 0.0006 |
| RHD*03.09 RHD*DIII.09 | RHD*186T, 410T, 455C, 667G | 1 | 0.0006 |
| RHD*05.01 RHD*DV.1 | RHD*667G,697C | 1 | 0.0006 |
| RHD*05.05 RHD*DV.5 | RHD*697A | 1 | 0.0006 |
| RHD*09.02.01 RHD*DAR2.01 | RHD*602G, 557G, 697C, 744T, 957A | 1 | 0.0006 |
| RHD*09.03 RHD*DAR3 | RHD*602G,667G | 1 | 0.0006 |
| RHD*10.00.02 RHD*DAU0.02 | RHD*1136T,150C | 1 | 0.0006 |
| No ISBT designation | RHD*1048C (GenBank: KC311353.1)† | 1 | 0.0006 |
| No ISBT designation | RHD*178C (GenBank: KX352163.1)† | 1 | 0.0006 |
| No ISBT designation | RHD*186T (GenBank: JN635688.1)† | 1 | 0.0006 |
| RHD*35 RHD*DMA | RHD*621C | 1 | 0.0006 |
| RHD*37 RHD*DUC2 | RHD*733C | 1 | 0.0006 |
| No ISBT designation | RHD*602G,733C,744T,1136T‡ | 1 | 0.0006 |
| No ISBT designation | RHD*648C (GenBank: JQ405073.1)† | 1 | 0.0006 |
| No ISBT designation | RHD*674T (GenBank: AF510070.1)† | 1 | 0.0006 |
| RHCE alleles | |||
| RHCE*01.01 RHCE*ce.01 | RHCE*ce48C | 402 | 0.2274 |
| RHCE*01 or RHCE*ce RHCE*c RHCE*e | RHCE*ce | 356 | 0.2014 |
| RHCE*01.20.01 RHCE*ce.20.01 RHCE*ceVS01 | RHCE*ce733G | 245 | 0.1386 |
| RHCE*02 or RHCE*Ce RHCE*C RHCE*e | RHCE*Ce | 207 | 0.1171 |
| RHCE*03 or RHCE*cE RHCE*c RHCE*E | RHCE*cE | 180 | 0.1018 |
| RHCE*01.20.03 RHCE*ce.20.03 RHCE*ceVS03 | RHCE*ceS | 80 | 0.0452 |
| RHCE*01.06.01 RHCE*ce.06.01 | RHCE*ceAG | 72 | 0.0407 |
| RHCE*01.20.02 RHCE*ce.20.02 RHCE*ceVS02 | RHCE*ce48C, 733G | 55 | 0.0311 |
| No ISBT designation (proposed as RHCE*01.01.02 RHCE*ce.01.02 | RHCE*ce48C,105T* | 53 | 0.0300 |
| RHCE*01.20.09 RHCE*ce.20.09 RHCE*ceVS09 | RHCE*ce48C, 733G, 941C | 47 | 0.0266 |
| RHCE*01.02.01 RHCE*ce.02.01 | RHCE*ceTI | 22 | 0.0124 |
| RHCE*01.07.01 RHCE*ce.07.01 | RHCE*ceMO | 20 | 0.0113 |
| RHCE*01.08 RHCE*ce.08 | RHCE*ceBI | 4 | 0.0023 |
| RHCE*01.20.06 RHCE*ce.20.06 RHCE*ceVS06 | RHCE*ceCF | 4 | 0.0023 |
| RHCE*01.05.01 RHCE*ce.05.01 | RHCE*ceEK | 3 | 0.0017 |
| RHCE*01.20.05 RHCE*ce.20.05 RHCE*ceVS05 | RHCE*ce733G, 1006T | 3 | 0.0017 |
| RHCE*03.04 RHCE*cE.04 | RHCE*cEIV | 3 | 0.0017 |
| RHCE*04 RHCE*CE | RHCE*CE | 2 | 0.0011 |
| RHCE*01.03 RHCE*ce.03 | RHCE*ce1025T | 1 | 0.0006 |
| RHCE*01.04.01 RHCE*ce.04.01 | RHCE*ceAR | 1 | 0.0006 |
| RHCE*01.06.02 RHCE*ce.06.02 | RHDE*ceAG.0 2 | 1 | 0.0006 |
| RHCE*01.06.05 RHCE*ce.06.05 | RHCE*ceAG.0 5 | 1 | 0.0006 |
| RHCE*01.20.04.02 RHCE*ce.20.04.02 RHCE*ceVS04.02 | RHCE*ceTI type 2-like | 1 | 0.0006 |
| RHCE*01.20.07 RHCE*ce.20.07 RHCE*ceVS07 | RHCE*ceJAL | 1 | 0.0006 |
| RHCE*02.08.01 RHCE*Ce.08.01 | RHCE*CeCW | 1 | 0.0006 |
| RHCE*02.22 RHCE*Ce.22 | RHCE*Ce667T | 1 | 0.0006 |
| RHCE*02.30 RHCE*Ce.30 | RHCE*Ce733G | 1 | 0.0006 |
| RHCE*03.18 RHCE*cE.18 | RHCE*cE48C | 1 | 0.0006 |
| Allele ISBT name | Allele detail/alias | Allele, n | Allele frequency |
|---|---|---|---|
| RHD alleles | |||
| RHD*01 | — | 920 | 0.5204 |
| RHD Deletion | — | 294 | 0.1663 |
| RHD*10.00 RHD*DAU0 | RHD*1136T | 229 | 0.1295 |
| RHD*08N.01RHD*Pseudo- gene RHD*Ψ | — | 66 | 0.0373 |
| RHD*03N.01 | RHD*DIIIa-CEVS (4-7)-D | 65 | 0.0368 |
| RHD*09.03.01 RHD*DAR3.01 | RHD*602G,667G | 57 | 0.0322 |
| RHD*10.03 RHD*DAU3 | RHD*835A,1136T | 35 | 0.0198 |
| RHD*10.00.01 RHD*DAU0.01 | RHD*1136T,579A | 21 | 0.0119 |
| RHD*04.01 RHD*DIVa | RHD*186T,410T, 455C,1048C | 16 | 0.0090 |
| RHD*10.05 RHD*DAU5 | RHD*667G,697C,1136T | 12 | 0.0068 |
| RHD*03.01 RHD*DIIIa | RHD*186T,410T,455C,602G,667G,819A | 10 | 0.0057 |
| No ISBT designation (proposed as RHD*03.01.02 RHD*DIII.01.02) | RHD*186T,410T,455C,602G,667G* | 7 | 0.0040 |
| RHD*01W.33RHD*weak D type 33 | RHD*520A | 3 | 0.0017 |
| RHD*09.01.02 RHD*DAR1.02 | RHD*602G,667G, 1025C | 3 | 0.0017 |
| No ISBT designation | RHD*841C (GenBank: KU363613.2)† | 3 | 0.0017 |
| RHD*01.01 | RHD*48C | 2 | 0.0011 |
| RHD*01W.66RHD*weak D type 66 | RHD*916A | 2 | 0.0011 |
| RHD*12.01 RHD*DOL1 | RHD*509C,667G | 2 | 0.0011 |
| RHD*49 RHD*DWN | RHD*1053T, 1057T, 1059G, 1060A, 1061A | 2 | 0.0011 |
| RHD* 01W.137RHD*weak D type 137 | RHD*780A | 1 | 0.0006 |
| RHD*01N.07 | RHD*D-CE (4-7)-D | 1 | 0.0006 |
| RHD*01N.35 | RHD*330_331delGT | 1 | 0.0006 |
| RHD*01W.1RHD*weak D type 1 | RHD*809G | 1 | 0.0006 |
| RHD*01W.45RHD*weak D type 45 | RHD*1195A | 1 | 0.0006 |
| RHD*03.09 RHD*DIII.09 | RHD*186T, 410T, 455C, 667G | 1 | 0.0006 |
| RHD*05.01 RHD*DV.1 | RHD*667G,697C | 1 | 0.0006 |
| RHD*05.05 RHD*DV.5 | RHD*697A | 1 | 0.0006 |
| RHD*09.02.01 RHD*DAR2.01 | RHD*602G, 557G, 697C, 744T, 957A | 1 | 0.0006 |
| RHD*09.03 RHD*DAR3 | RHD*602G,667G | 1 | 0.0006 |
| RHD*10.00.02 RHD*DAU0.02 | RHD*1136T,150C | 1 | 0.0006 |
| No ISBT designation | RHD*1048C (GenBank: KC311353.1)† | 1 | 0.0006 |
| No ISBT designation | RHD*178C (GenBank: KX352163.1)† | 1 | 0.0006 |
| No ISBT designation | RHD*186T (GenBank: JN635688.1)† | 1 | 0.0006 |
| RHD*35 RHD*DMA | RHD*621C | 1 | 0.0006 |
| RHD*37 RHD*DUC2 | RHD*733C | 1 | 0.0006 |
| No ISBT designation | RHD*602G,733C,744T,1136T‡ | 1 | 0.0006 |
| No ISBT designation | RHD*648C (GenBank: JQ405073.1)† | 1 | 0.0006 |
| No ISBT designation | RHD*674T (GenBank: AF510070.1)† | 1 | 0.0006 |
| RHCE alleles | |||
| RHCE*01.01 RHCE*ce.01 | RHCE*ce48C | 402 | 0.2274 |
| RHCE*01 or RHCE*ce RHCE*c RHCE*e | RHCE*ce | 356 | 0.2014 |
| RHCE*01.20.01 RHCE*ce.20.01 RHCE*ceVS01 | RHCE*ce733G | 245 | 0.1386 |
| RHCE*02 or RHCE*Ce RHCE*C RHCE*e | RHCE*Ce | 207 | 0.1171 |
| RHCE*03 or RHCE*cE RHCE*c RHCE*E | RHCE*cE | 180 | 0.1018 |
| RHCE*01.20.03 RHCE*ce.20.03 RHCE*ceVS03 | RHCE*ceS | 80 | 0.0452 |
| RHCE*01.06.01 RHCE*ce.06.01 | RHCE*ceAG | 72 | 0.0407 |
| RHCE*01.20.02 RHCE*ce.20.02 RHCE*ceVS02 | RHCE*ce48C, 733G | 55 | 0.0311 |
| No ISBT designation (proposed as RHCE*01.01.02 RHCE*ce.01.02 | RHCE*ce48C,105T* | 53 | 0.0300 |
| RHCE*01.20.09 RHCE*ce.20.09 RHCE*ceVS09 | RHCE*ce48C, 733G, 941C | 47 | 0.0266 |
| RHCE*01.02.01 RHCE*ce.02.01 | RHCE*ceTI | 22 | 0.0124 |
| RHCE*01.07.01 RHCE*ce.07.01 | RHCE*ceMO | 20 | 0.0113 |
| RHCE*01.08 RHCE*ce.08 | RHCE*ceBI | 4 | 0.0023 |
| RHCE*01.20.06 RHCE*ce.20.06 RHCE*ceVS06 | RHCE*ceCF | 4 | 0.0023 |
| RHCE*01.05.01 RHCE*ce.05.01 | RHCE*ceEK | 3 | 0.0017 |
| RHCE*01.20.05 RHCE*ce.20.05 RHCE*ceVS05 | RHCE*ce733G, 1006T | 3 | 0.0017 |
| RHCE*03.04 RHCE*cE.04 | RHCE*cEIV | 3 | 0.0017 |
| RHCE*04 RHCE*CE | RHCE*CE | 2 | 0.0011 |
| RHCE*01.03 RHCE*ce.03 | RHCE*ce1025T | 1 | 0.0006 |
| RHCE*01.04.01 RHCE*ce.04.01 | RHCE*ceAR | 1 | 0.0006 |
| RHCE*01.06.02 RHCE*ce.06.02 | RHDE*ceAG.0 2 | 1 | 0.0006 |
| RHCE*01.06.05 RHCE*ce.06.05 | RHCE*ceAG.0 5 | 1 | 0.0006 |
| RHCE*01.20.04.02 RHCE*ce.20.04.02 RHCE*ceVS04.02 | RHCE*ceTI type 2-like | 1 | 0.0006 |
| RHCE*01.20.07 RHCE*ce.20.07 RHCE*ceVS07 | RHCE*ceJAL | 1 | 0.0006 |
| RHCE*02.08.01 RHCE*Ce.08.01 | RHCE*CeCW | 1 | 0.0006 |
| RHCE*02.22 RHCE*Ce.22 | RHCE*Ce667T | 1 | 0.0006 |
| RHCE*02.30 RHCE*Ce.30 | RHCE*Ce733G | 1 | 0.0006 |
| RHCE*03.18 RHCE*cE.18 | RHCE*cE48C | 1 | 0.0006 |
Alleles that are not considered by ISBT because of identical phenotype changes to alleles included in ISBT. RHD*186T,410T,455C,602G,667G has the same amino acid changes as RHD*03.01 RHD*DIIIa (RHD*186T,410T,455C,602G,667G,819A), with c. 819G>A being silent; RHCE*ce48C,105T has the same amino-acid changes as RHCE*48C, with c. 105C>T being silent. Proposed designations in line with the anticipated ISBT nomenclature for the 2 alleles are included in parentheses.
Alleles that are not included by ISBT but have been reported to GenBank. GenBank IDs are listed.
Novel RHD allele.
![Determination of RHD zygosity and RH hybrid alleles, by using sequencing coverage by RHtyper. (A) RHtyper determined RHD homozygous and hemizygous deletions by comparing the sequencing coverage across the entire RHD region with the average coverage of the whole genome. The distribution of log2-transformed coverage ratios of RHD and RHCE regions of samples SJSCD040770 (no RHD deletion), SJSCD040771 (hemizygous RHD deletion), and SJSCD040777 (homozygous RHD deletion) are shown. Blue and red dotted lines represent the cutoff for the hemizygous (−0.6) and homozygous (−3) deletions, respectively. (B) RHtyper predicted hybrid alleles by their characteristic SNPs and indels supplemented by sequencing coverage profiling. The potential boundaries of the hybrid alleles are predicted by the CBS algorithm, using sequencing coverage. The distribution of log2-transformed coverage ratios of RHD and RHCE genes and the potential hybrid boundaries of sample SJSCD040934 [RHD/RHD*D-CE(4-7)-D] are shown. Each dot represents a 300-bp segment. Segments of introns are indicated by blue dots, and segments of exons are indicated by colors other than blue. The boundaries of the hybrid allele are indicated by vertical gray lines with the genomic location of the potential breakpoints labeled.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/18/10.1182_bloodadvances.2020002148/2/m_advancesadv2020002148f3.png?Expires=1765939609&Signature=fklHRMBP60FQJJUGrZJYZcXfCMP5bf-IzQdDllqhESYcwdC3zWO0Iyg~j9sRS-KiPjERkMDan4heex1S-28uc0fDuNVHuQhFahxFBNK~nMRfgfeFE6znjp9Q2wtvDEvamS4r6UdFPstzYhshjpMpIzRld9FQr8k5EDbKgsqIR0cgNKwzjNplpCKsZq0WyfZwI1gzrgTozfJnaebQRCsyOOXnX-~y3NHQqkDp0A2Jpyf-FwqgEzrWHe4f8eQWTxT2WOsc1n5uJce~BJGDAYBcB-6E~6yRumQiQu~Cz6~Pb1z5H0Oehj57Yiq5HAUvXuTgNXmB-iT-odW4YI3qYcb9fw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Determination of RHD zygosity and RH hybrid alleles, by using sequencing coverage by RHtyper. (A) RHtyper determined RHD homozygous and hemizygous deletions by comparing the sequencing coverage across the entire RHD region with the average coverage of the whole genome. The distribution of log2-transformed coverage ratios of RHD and RHCE regions of samples SJSCD040770 (no RHD deletion), SJSCD040771 (hemizygous RHD deletion), and SJSCD040777 (homozygous RHD deletion) are shown. Blue and red dotted lines represent the cutoff for the hemizygous (−0.6) and homozygous (−3) deletions, respectively. (B) RHtyper predicted hybrid alleles by their characteristic SNPs and indels supplemented by sequencing coverage profiling. The potential boundaries of the hybrid alleles are predicted by the CBS algorithm, using sequencing coverage. The distribution of log2-transformed coverage ratios of RHD and RHCE genes and the potential hybrid boundaries of sample SJSCD040934 [RHD/RHD*D-CE(4-7)-D] are shown. Each dot represents a 300-bp segment. Segments of introns are indicated by blue dots, and segments of exons are indicated by colors other than blue. The boundaries of the hybrid allele are indicated by vertical gray lines with the genomic location of the potential breakpoints labeled.
Determination of RHD zygosity and RH hybrid alleles, by using sequencing coverage by RHtyper. (A) RHtyper determined RHD homozygous and hemizygous deletions by comparing the sequencing coverage across the entire RHD region with the average coverage of the whole genome. The distribution of log2-transformed coverage ratios of RHD and RHCE regions of samples SJSCD040770 (no RHD deletion), SJSCD040771 (hemizygous RHD deletion), and SJSCD040777 (homozygous RHD deletion) are shown. Blue and red dotted lines represent the cutoff for the hemizygous (−0.6) and homozygous (−3) deletions, respectively. (B) RHtyper predicted hybrid alleles by their characteristic SNPs and indels supplemented by sequencing coverage profiling. The potential boundaries of the hybrid alleles are predicted by the CBS algorithm, using sequencing coverage. The distribution of log2-transformed coverage ratios of RHD and RHCE genes and the potential hybrid boundaries of sample SJSCD040934 [RHD/RHD*D-CE(4-7)-D] are shown. Each dot represents a 300-bp segment. Segments of introns are indicated by blue dots, and segments of exons are indicated by colors other than blue. The boundaries of the hybrid allele are indicated by vertical gray lines with the genomic location of the potential breakpoints labeled.
In the 884-patient cohort, RHtyper identified 28 distinct RHCE alleles (Table 3). The 3 most frequently present RHCE alleles were RHCE*ce48C (n = 402; 0.2274), RHCE*ce (n = 356; 0.2014), and RHCE*ce733G (n = 245; 0.1386), consistent with previous findings in individuals of African descent.2,3 To phase SNPs (ie, SNPs on the same allele or on 2 different alleles) that are separated by thousands of base pairs, RHtyper imputed SNP and indel haplotype associations and RHD/RHCE linkages based on observations in individuals of African descent (https://www.isbtweb.org) and published allele frequencies.2 For samples containing RHCE*ce with c.48G/C and c.733C/G only, RHtyper phased c.48C and c.733G, considering RHD/RHCE linkages according to prevalence determined by RH mRNA sequencing results (unpublished data). The C antigen is encoded by RHCE*C alleles, and genomic identification of RHCE*C requires additional considerations.11,12,15 RHtyper identified RHCE*C by c.48C in RHCE*C exon 1 and a 109-bp insertion in RHCE*C intron 2. In 360 samples with historical serologic C typing available, RHtyper achieved a concordance of 98.3% (354 of 360 samples). Among the 6 discordant samples, RHCE*ceTI, when in cis to RHD*DIVa, can result in false-positive serologic C typing (sample SJSCD045321 and SJSD045346; Table 4).31 Of note, although the hybrid allele RHD*DIIIa-CEVS(4–7)-D encodes a partial C antigen from the RHD locus and typically types positive with monoclonal anti-C reagents,32 serologic C typing for sample SJSCD043391 and SJSCD045316 was recorded as negative.
Discordance between predicted phenotype and serology results of the C antigen
| Patient ID | RHCE | RHD | Predicated phenotype | Serology result | ||
|---|---|---|---|---|---|---|
| Allele1 | Allele2 | Allele 1 | Allele 2 | |||
| SJSCD045321* | RHCE*ceAG | RHCE*ceTI | RHD Deletion | RHD*DIVa | C neg | C pos |
| SJSCD045346* | RHCE*ceTI | RHCE*ce48C | RHD*DIVa | RHD*DAU0 | C neg | C pos |
| SJSCD043391 | RHCE*ce733G, 1006T | RHCE*Ce733G | RHD*DAR3.01 | RHD*DIIIa-CEVS(4- 7)-D | C pos | C neg |
| SJSCD045316 | RHce(48C,105T) | RHCE*ceS | RHD Deletion | RHD*DIIIa-CEVS(4- 7)-D | C pos | C neg |
| SJSCD043189 | RHCE*ce48C | RHCE*ce48C | RHD*DV.1 | RHD*DAU3 | C neg | C pos |
| SJSCD043291 | RHCE*ceS | RHCE*ceCF | RHD*DIIIa | RHD*DAR3 | C neg | C pos |
| Patient ID | RHCE | RHD | Predicated phenotype | Serology result | ||
|---|---|---|---|---|---|---|
| Allele1 | Allele2 | Allele 1 | Allele 2 | |||
| SJSCD045321* | RHCE*ceAG | RHCE*ceTI | RHD Deletion | RHD*DIVa | C neg | C pos |
| SJSCD045346* | RHCE*ceTI | RHCE*ce48C | RHD*DIVa | RHD*DAU0 | C neg | C pos |
| SJSCD043391 | RHCE*ce733G, 1006T | RHCE*Ce733G | RHD*DAR3.01 | RHD*DIIIa-CEVS(4- 7)-D | C pos | C neg |
| SJSCD045316 | RHce(48C,105T) | RHCE*ceS | RHD Deletion | RHD*DIIIa-CEVS(4- 7)-D | C pos | C neg |
| SJSCD043189 | RHCE*ce48C | RHCE*ce48C | RHD*DV.1 | RHD*DAU3 | C neg | C pos |
| SJSCD043291 | RHCE*ceS | RHCE*ceCF | RHD*DIIIa | RHD*DAR3 | C neg | C pos |
RHCE*ceTI, when in cis to RHD*DIVa, can result in false-positive typing with monoclonal anti-C antibodies.
RHtyper identified 28 samples with RH alleles containing additional novel or low-frequency SNPs. Low-frequency SNPs were found in ≤5 samples in the 884 patients (supplemental Table 2). By Sanger sequencing, we confirmed 1 unreported RHD allele that appears to belong to the RHD DAU family (RHD*c.1136T with c. 602G, 733C, 744T; SJSCD040786). One unreported RHCE allele (RHCE*ce48C with c.520A; SJSCD045681) was also found, but no remaining sample was available for confirmation. For the other samples, either the additional SNPs were silent or it could not be phased because of the distance between SNPs. The closest known RH allele was assigned to samples with additional SNPs that were silent, could not be phased, or could not be confirmed because samples were not available (supplemental Table 3A-B).
We performed further confirmation of the identified SNPs and indels in the 884-patient cohort (supplemental Methods; supplemental Figure 2). Available WES data from 402 patients were used to verify 100% (603 of 603) of RHD and 99.86% (707 of 708) of RHCE exonic SNPs and indels detected by WGS. An SNP in 1 sample was not detected because of low WES coverage (<3 supporting WES reads). RHtyper identified 20 distinct low-frequency RHD SNPs and 13 distinct low-frequency RHCE SNPs (supplemental Table 2). We confirmed these by Sanger sequencing in all available samples (37 of 40 low-frequency RHD SNP samples, and 25 of 30 low-frequency RHCE SNP samples).
Discussion
RH genotyping by NGS methods can be used to determine the presence of altered alleles,11,33-37 but the time-consuming analysis and subject matter expertise required for data interpretation limit clinical implementation. To simplify and automate the process, we developed RHtyper, which uses a Bayesian likelihood-based framework to infer RH genotypes directly after sequence read alignment. The algorithm considers both sequence consistency at each SNP and indel and phase consistency across adjacent SNPs and indels to improve prediction accuracy. RHtyper incorporates coverage profiling to determine RHD zygosity, characteristic SNPs and coverage profiling to identify hybrid alleles, and the CBS algorithm to define the potential boundaries of the hybrid alleles. In a validation cohort consisting of 57 patients with SCD, RHtyper achieved 100% accuracy for RHD and 98.2% accuracy for RHCE, when compared with genotypes obtained by RH BeadChip and targeted molecular assays and after verification by Sanger sequencing and independent NGS assays. Subsequent implementation of RHtyper to analyze WGS data from 884 patients identified a novel RHD DAU family allele (RHD*602G, 733C, 744T, 1136T).
Several published algorithms for automatic RBC antigen typing with NGS data are available but have not been tested on a large number of individuals of African descent, in whom RH genes are significantly more heterogeneous. An algorithm termed BOOGIE genotyped ABO and RHD by using hidden Markov models.16 From 69 individuals in the Personal Genome Project, this algorithm achieved a concordance of 94% for predicting RhD serologic type. To determine the best allele pairs, BOOGIE uses a 1-nearest neighbor algorithm that relies heavily on accurate SNP-calling results from third-party tools. Lane et al used WGS data to develop a rule-based algorithm for RBC and platelet antigen typing.12 In 110 MedSeq Project participants, the final algorithm was 99.8% concordant with phenotypes typed by serology and SNP assays. Notably, most of the participants were white Americans, and only 11.8% (13 of 110) were African Americans; common Rh phenotypes, not RH genotypes, were analyzed. Similar to BOOGIE, this algorithm relies on multiple external tools, including the Genome Analysis Toolkit and BEDtools, to obtain SNP and sequencing coverage information. Therefore, misalignments occurring in the external tools may be ignored and result in incorrect genotyping. RHtyper is a stand-alone algorithm that does not need external support tools. Misalignments can be discovered by scrutinizing at the read level and prevented by adjusting the algorithm. Furthermore, RHtyper is optimized to determine the complex RH genotypes of individuals of African descent and can be readily updated with newly identified RH alleles and haplotype associations. It can also be tailored toward various racial and ethnic populations by modifying the allele ranking mechanism. RHtyper predicts RH genotypes based on SNPs and indels found mostly in RH exons, and thus WES data can be used to genotype most RH alleles. However, alleles that depend on intron markers for accurate interpretation, most notably RHCE*C (by c.48C in RHCE*C exon 1 and a 109-bp insertion in RHCE*C intron 2), cannot be detected by RHtyper when using WES data alone, currently. We have implemented RHtyper as a cloud-based public access application in DNAnexus (https://platform.dnanexus.com/app/RHtyper), permitting clinicians and researchers to perform WGS-based RH genotyping.
Alloimmunization, particularly to the Rh system, is one of the most common complications in patients with SCD who are receiving transfusions. Alloantibodies increase the risk of hemolytic transfusion reactions and reduce the number of compatible RBC units, thus affecting the overall care and survival of patients with SCD.38-41 Although currently cost prohibitive, it is feasible to provide RH genotype–matched transfusions if a robust African American donor pool is available.2 As the cost of NGS continues to decrease, affordable genome sequencing for patients with chronic diseases will soon be possible. Using existing genome sequencing data could be a cost-effective and comprehensive approach for obtaining RH and other blood group genotypes.42 A major barrier to this goal can be overcome by our analytical approaches, with enhanced ability to distinguish the extensive genetic RH variation in individuals of African descent and with improved capacity to conduct large-scale, population-level analyses.
A limitation of this study is the standard short-read WGS data used. Because RHD and RHCE are a duplicated gene family and standard WGS-generated, short-length sequence reads are of 150 to 200 bp, sequence read misalignment between the 2 RH genes can occur, resulting in erroneous genotypes. Another remaining challenge with short-length sequence reads is the ability to phase SNPs that are located thousands of base pairs apart. In this study, we relied on the known haplotype associations and allele frequencies among individuals of African descent for phase prediction. However, this approach cannot be applied to rare and novel SNPs. These bioinformatics challenges can be overcome by long-read WGS. Single-molecule, real-time sequencing technology and nanopore technology can generate extra-long sequence reads to improve sequence read alignment and SNP phasing. Currently, long-read WGS is more expensive and has a much higher error rate than standard WGS; further improvement is needed. RHtyper can be adjusted to suit long-read WGS platforms. Long-read WGS uses a different base quality value for sequencing reads; therefore, the likelihood score calculation for allele pair prediction should be modified. Also, current long-read WGS is prone to erroneous variant calling and inaccurate indels, which must be considered when adapting the RHtyper algorithm to long-read WGS platforms. Another limitation of the study is that validation of RHtyper with full serologic Rh phenotypes was not performed. Among 360 samples with historical serologic C typing available, RHtyper achieved 98.3% (354 of 360) concordance for predicting the C antigen. Although 2 samples with RHCE*ceTI in cis to RHD*DIVa are associated with false-positive serologic C typing, the remaining 4 discrepancies are most likely caused by sample mislabeling and errors in manual testing, interpretation, and recording.43 Our goal was to develop and implement an RH genotyping algorithm for a large cohort of patients with SCD and correlate our findings with known RH allele frequency.
We have developed a computational algorithm, RHtyper, for comprehensive and high-throughput RH genotyping from WGS data. This approach can identify diverse RH genetic variation present in patients with SCD and can be implemented in those with existing WGS data. Knowledge of individual RH genotype currently aids antibody identification and selection of donor units for Rh-alloimmunized patients. More research is necessary to determine the immunogenicity of specific RH variant alleles to further refine red cell matching by genotype for patients with SCD.
The original WGS data from 884 patients with SCD are available at St Jude Cloud (https://platform.stjude.cloud/requests/cohorts; accession number: SJC-DS-1006).29
The RH genotypes of the 884 patients are included as supplemental data. We have also implemented RHtyper as a cloud-based public access application in DNAnexus (https://platform.dnanexus.com/app/RHtyper), allowing for NGS-based RH genotyping. A link to RHtyper tutorial and readme can also be found on the Web site.
Acknowledgments
The authors thank the SCCRIP study team, the Biorepository, and the Hartwell Center at St Jude for sharing WGS data, providing patient samples for confirmation, and performing Sanger sequencing, respectively.
This study was supported by National Institutes of Health (NIH), National Cancer Institute grant CA021765; the American Lebanese Syrian Associated Charities (Y.Z.); National Blood Foundation Early-Stage Investigator’s Award (Y.Z.); and NIH, National Heart, Lung, and Blood Institute grant HL147879-01 (S.T.C. and C.M.W.).
Authorship
Contribution: J.S.H., M.J.W., C.M.W., S.T.C., and Y.Z. designed the research; T.-C.C. and G.W. designed and developed the RHtyper; K.M.H., J.Y., and S.V. performed confirmatory experiments; E.R., V.A.S., J.M.F., J.S.H., and M.J.W. provided patient samples and WGS data; and T.-C.C., C.M.W., S.T.C., and Y.Z. wrote the manuscript.
Conflict-of-interest disclosure: M.J.W. holds equity in Beam Therapeutics, Rubius Inc, Bristol Myers Squibb, Roche, Cellarity Inc, and Incyte, and also receives consulting fees from Novartis and Esperion Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Yan Zheng, Department of Pathology, St Jude Children's Research Hospital, 262 Danny Thomas Pl, MS-342, Memphis, TN 38105; e-mail: yan.zheng@stjude.org.

