


Abstract
Long-term cure of childhood Burkitt lymphoma (BL) in sub-Saharan Africa after treatment with single-agent cyclophosphamide has been documented for more than half of a century. Contemporary cure rates for the highest-risk patients with BL in high-income countries exceed 90% using intensive multiagent chemotherapy. By contrast, the majority of African children with BL still die. Data spanning 5 decades in Africa have repeatedly shown that the children most likely to achieve cure with limited cyclophosphamide regimens are those with lower-stage disease isolated to the jaw. Attempts to intensify the cyclophosphamide monotherapy backbone with the addition of vincristine, low-dose methotrexate, prednisone, doxorubicin, and/or low-dose cytarabine have not yielded significant improvement. High-dose methotrexate is a critical component in the treatment of childhood BL worldwide. Although initial efforts in Africa to incorporate high-dose methotrexate resulted in high treatment-related mortality, more recent collaborative experiences from North and West Africa, as well as Central America, demonstrate that it can be administered safely and effectively, despite limitations in supportive care resources. Recognizing the unacceptable disparity in curative outcomes for BL between the United States/Europe and equatorial Africa, there is a critical need to safely adapt contemporary treatment regimens to optimize curative outcomes amid the resource limitations in regions where BL is endemic. Here, we critically review reports of BL treatment outcomes from low- and middle-income countries, in addition to data from high-income countries that predated modern intensified regimens, to identify potential strategies to improve the therapeutic approach for children suffering from BL in sub-Saharan Africa.
Introduction
The history of Burkitt lymphoma (BL) offers a snapshot into the pervasive survival disparity for childhood cancer between high-income countries (HICs) and the rest of the world.1 After its description by Denis Burkitt and colleagues in Uganda in the 1960s, BL became recognized as the most common non-Hodgkin lymphoma (NHL) of childhood worldwide.2,3 Considered the most common overall pediatric cancer in regions of sub-Saharan Africa (SSA) where malaria is endemic, “endemic” BL has captivated clinicians and scientists alike for more than half a century. Tumor biopsies from African children with BL have revealed fundamental mechanisms of cancer biology, from the original discovery of Epstein-Barr virus and the c-myc oncogene, to determination of the genomic signature of BL.4-8 Parallel to the biological discoveries related to BL, pediatric cancer cooperative group trials in HICs have steadily improved the outcomes of children with BL by generating evidence for improving diagnostic accuracy, staging classification, risk stratification, supportive care, and risk-adapted chemoimmunotherapy. The impact of progressive cooperative group trials is highlighted, in part, by contemporary pediatric BL cure rates that exceed 90% in HICs.9-16 However, advances in HICs are not directly applicable to the African health care landscape, and, putatively, to the biology of endemic BL. Therefore, the majority of children with BL worldwide have not benefited from these advances. Best estimates of long-term survival of pediatric BL in SSA today are 30% to 50%, which are unchanged from the 1970s era, the discovery of BL and the dawn of the field of pediatric oncology. To identify strategies to accelerate cure rates for children with endemic BL, we critically review the evolution of pediatric BL treatments and outcomes from trials and retrospective clinical reports in SSA, in parallel with developments in HICs.
Context of BL treatment and outcome studies in SSA
Interpretation of reports describing clinical outcomes of BL in SSA must take into account the heterogeneity of diagnostic and staging tools, timing and dosing of the intended chemotherapy drugs, response criteria, and duration of follow-up. These factors are dictated by available infrastructure for care and clinical research and the paucity of established cooperative groups to enable multicenter trials. Significant variability might exist between treating centers, for different children within a given treatment center, and for the same patient during a treatment course. For example, many centers in SSA diagnose BL based on clinical presentation only or tumor morphology alone.17 These modalities have been shown to have limited diagnostic accuracy.18,19 Moreover, staging of patients with BL requires routine microscopy of cerebrospinal fluid (CSF) samples and bone marrow evaluation to define extent of disease; however, most centers in SSA lack resources to consistently perform these staging tests. Similarly, assessment of treatment response relying solely on symptoms and clinical examination may miss subtle tumors, and it has not been standardized across centers or between clinicians. Lastly, the majority of data are limited to retrospective review of records, which is impacted by access to records, accuracy of clinical information, and potential underrepresentation of dead and lost patients.20 Acknowledging these limitations, review of studies from SSA is valuable to demonstrate prevailing practices, as well as trends and gaps in patient outcomes, which can inform application of evidence from HICs.
The quality and speed of diagnostic and staging processes have critical implications in long-term outcomes for patients. Diagnostic delays can consequently lead to patients having a greater burden of disease and more complex clinical scenarios (eg, higher risk for tumor lysis syndrome or more severe neurologic complications from spinal cord compression/cranial nerve palsies). Widespread societal poverty has detrimental impact on access to care, often leading to delays in diagnosis; the financial resources required for long-distance transportation to tertiary care medical facilities can be prohibitive and a major hurdle to overcome. Additionally, diagnostic delays within the medical system are important to consider. Patients often present to medical care initially at smaller rural health care centers where, aside from the dramatic and conspicuous presentation of a jaw tumor, there is lack of awareness or recognition of other clinical characteristics of BL, such as an abdominal mass. This, coupled with the challenge of accessing referral networks to the limited number of quaternary-level medical facilities that provide diagnostic and therapeutic resources for childhood cancer, lead to further delays in diagnosis. In efforts to overcome these limitations, international collaborations have focused efforts to bolster diagnostic accuracy and efficiency through telepathology portals.21-23 These types of global partnerships have the potential to improve accuracy of diagnosis and treatment through knowledge transfer between institutions in SSA and HICs. Enhancing the local capacity of the health care workforce and creating programs to train pediatric hematology/oncology experts are also essential to deliver the complex care required to support the intense therapy that is necessary to cure BL.24
Historical use of cyclophosphamide monotherapy for endemic BL
Treatment of BL at the Uganda Cancer Institute in the 1960s and 1970s represented some of the earliest experiences with chemotherapy in cancer treatment worldwide.25 Denis Burkitt and his contemporaries showed that even a single dose of cyclophosphamide monotherapy could induce clinical remission in 70% to 80% of patients with BL.26,27 However, only ∼25% of these patients had sustained remission, even after multiple doses of cyclophosphamide were administered.2,26,27 It became apparent that patients with advanced-stage disease suffered significantly higher rates of relapse and death.25,26,28 Current understanding of the pharmacodynamics of anticancer chemotherapy recognizes that cancer cells almost inevitably develop resistance to cytotoxic agents through multifaceted mechanisms. Specific to cyclophosphamide, resistance may involve decreased activation by liver cytochromes, decreased entry into or increased exit from tumor cells, increased deactivation inside tumor cells, increased cellular thiol level, increased activity of glutathione S-transferases, increased aldehyde dehydrogenase (ALDH1A and ALDH3) activity, and increased DNA repair capacity.29,30 Combination chemotherapy is a key strategy to overcome tumor resistance to cytotoxic agents.31 In addition to the cytotoxic effect of cyclophosphamide, it has an immunomodulatory effect that may play a role in effectiveness against lymphoid malignancies.32
Historical cyclophosphamide-based combination therapy for endemic BL
The incorporation of multiple low-intensity chemotherapeutic agents into the backbone of cyclophosphamide did not significantly improve long-term overall survival (OS) for endemic BL in the early years of the experience.25,27 Such regimens included cyclophosphamide, vincristine, methotrexate (MTX), and cytarabine, as well as cyclophosphamide, vincristine, and MTX (COM), both of which used low-dose MTX (ie, cumulative doses per cycle <100 mg/m2).25 Notably, these combinations were administered uniformly through the course of treatment, regardless of the risk of tumor lysis syndrome and with no induction and consolidation cycle variations. Although well tolerated, these low-intensity combinations were not superior to cyclophosphamide monotherapy.27 It was also still apparent that, like cyclophosphamide monotherapy, advanced-stage disease was associated with high rates of early relapse after treatment with these low-intensity regimens, including in the central nervous system (CNS).25,27
Early strategies to treat and prevent CNS relapse in SSA included the use of systemic agents purported to cross the blood-brain barrier, intrathecal administration of cytotoxic agents, and cranial radiotherapy. The addition of CNS-directed systemic chemotherapy in the form of lomustine delayed CNS relapses but was associated with increased mortality.33 Similarly, short-term intrathecal chemotherapy showed no efficacy in preventing CNS relapses.34 Borrowing from the experience of using cranial irradiation to prevent CNS relapse in children with acute lymphoblastic leukemia in HICs, Olweny et al conducted a trial of craniospinal irradiation vs no additional CNS-directed therapy in 22 children with BL following a CNS− remission after cyclophosphamide monotherapy or COM.35 In this small cohort, more CNS relapses (54%) were observed in the irradiated group compared with in the no-irradiation group (36%); ultimately, relapse frequency was found to correlate with advanced-stage disease. Subsequent studies from around the world showed severe long-term adverse effects of cranial irradiation in children; hence, attempts to use this modality in BL were abandoned.
Contemporary standards and efforts to improve BL treatment in SSA
Current standards of BL treatment in SSA are still primarily based on the successes of pioneering trials from the 1970s with cyclophosphamide as the backbone agent, without differentiation of staging or risk group.25,27,36-41 Survival rates have plateaued over the past 40 years, and efforts to improve have stalled since then. Contemporary efforts to improve outcomes within the cyclophosphamide-based paradigm have included the addition of various low doses of MTX,42-44 vincristine,45 doxorubicin, and prednisone.17,20,46,47 The collective experience has demonstrated cohort survival rates ranging from 30% to 50%, with mortality primarily attributed to tumor progression or recurrence (Table 1). Patients with stage I/II BL have achieved survival of 40% to 60%, whereas survival for stage III/IV BL (typically involving the abdominal cavity, bone marrow, or CNS) has remained <30%.26,38,40,41,48-50 This is particularly relevant because numerous studies in SSA have demonstrated that abdominal mass has become at least as common as jaw mass as a presenting feature of endemic BL (Figure 1).17,20,51,52
Survival patterns for pediatric BL treated with various chemotherapy regimens in SSA over 5 decades
| Location | Years | Sample size, n | Regimen | Chemotherapy dose, mg/m2 per cycle | Survival analysis | Stage III/IV survival, % | TRM, % | Cohort survival, % | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cyclophos | MTX | VCR | Doxo | Pred | |||||||||
| Uganda | 1967-1970 | 57 | Cyclophos | 1200 | — | — | — | — | OS | 24/0 | 18 | 30 | |
| Ghana | 1968-1972 | 110 | Cyclophos | 1200 | — | — | — | — | 2-y OS | 31/20 | NR | 44 | |
| Uganda | 1967-1977 | 280 | Various* | OS | 50/25 | NR | 39 | ||||||
| Lilongwe, Malawi | 1991-1997 | 73 | Cyclophos | 1200 | — | — | — | — | OS | 33 | NR | 55 | |
| Blantyre, Malawi | 2000-2002 | 42 | Plus hdMTX | 500 | 2000 | 2 | — | 420 | 1-y EFS | 24/25 | 33 | 33 | |
| GFAOP 3 | 2001-2004 | 187 | COPM | 1500 | 3000 | 1.5 | — | 300 | 1-y EFS | 52/36 | 21 | 56 | |
| GFAOP 6 | 2005-2008 | 178 | Cyclophos | 1200 | — | — | — | — | 1-y EFS | 30/16 | 8 | 33 | |
| Cameroon | 2006-2008 | 95 | Cyclophos | 1200-1800 | — | — | — | — | 1-y EFS | 33 | 7 | 35 | |
| Kenya | 2003-2011 | 428 | Plus doxo | 1200 | — | 1.5 | 60 | Tapering | 1-y EFS | NR | NR | 31 | |
| Blantyre, Malawi | 2010-2012 | 70 | Plus VCR | 1200-1800 | — | 1.5 | — | — | 1-y EFS | 24/32 | 3 | 48 | |
| Lilongwe, Malawi | 2011-2013 | 74 | CHOP-like | 800 | — | 1.5 | 40 | 300 | 18-mo OS | 26 | 8 | 29 | |
| GFAOP 7 | 2009-2015 | 400 | COPM | 1500 | 3000 | 1.5 | — | 420 | 1-y OS | 60/31 | 12 | 60 | |
| Lilongwe, Malawi | 2013-2015 | 73 | CHOP-like | 800 | — | 1.5 | 40 | 300 | 18-mo OS | 28/17 | 16 | 29 | |
| Uganda | 2012-2017 | 180 | COM | 900 | 15 | 1.5 | — | — | 4-y OS | 42 | NR | 44 | |
| Location | Years | Sample size, n | Regimen | Chemotherapy dose, mg/m2 per cycle | Survival analysis | Stage III/IV survival, % | TRM, % | Cohort survival, % | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cyclophos | MTX | VCR | Doxo | Pred | |||||||||
| Uganda | 1967-1970 | 57 | Cyclophos | 1200 | — | — | — | — | OS | 24/0 | 18 | 30 | |
| Ghana | 1968-1972 | 110 | Cyclophos | 1200 | — | — | — | — | 2-y OS | 31/20 | NR | 44 | |
| Uganda | 1967-1977 | 280 | Various* | OS | 50/25 | NR | 39 | ||||||
| Lilongwe, Malawi | 1991-1997 | 73 | Cyclophos | 1200 | — | — | — | — | OS | 33 | NR | 55 | |
| Blantyre, Malawi | 2000-2002 | 42 | Plus hdMTX | 500 | 2000 | 2 | — | 420 | 1-y EFS | 24/25 | 33 | 33 | |
| GFAOP 3 | 2001-2004 | 187 | COPM | 1500 | 3000 | 1.5 | — | 300 | 1-y EFS | 52/36 | 21 | 56 | |
| GFAOP 6 | 2005-2008 | 178 | Cyclophos | 1200 | — | — | — | — | 1-y EFS | 30/16 | 8 | 33 | |
| Cameroon | 2006-2008 | 95 | Cyclophos | 1200-1800 | — | — | — | — | 1-y EFS | 33 | 7 | 35 | |
| Kenya | 2003-2011 | 428 | Plus doxo | 1200 | — | 1.5 | 60 | Tapering | 1-y EFS | NR | NR | 31 | |
| Blantyre, Malawi | 2010-2012 | 70 | Plus VCR | 1200-1800 | — | 1.5 | — | — | 1-y EFS | 24/32 | 3 | 48 | |
| Lilongwe, Malawi | 2011-2013 | 74 | CHOP-like | 800 | — | 1.5 | 40 | 300 | 18-mo OS | 26 | 8 | 29 | |
| GFAOP 7 | 2009-2015 | 400 | COPM | 1500 | 3000 | 1.5 | — | 420 | 1-y OS | 60/31 | 12 | 60 | |
| Lilongwe, Malawi | 2013-2015 | 73 | CHOP-like | 800 | — | 1.5 | 40 | 300 | 18-mo OS | 28/17 | 16 | 29 | |
| Uganda | 2012-2017 | 180 | COM | 900 | 15 | 1.5 | — | — | 4-y OS | 42 | NR | 44 | |
Groupe Franco-Africain d’Oncologie Pédiatrique (GFAOP) 3 includes Cameroon, Madagascar, and Senegal; GFAOP 6 includes Cameroon, Madagascar, Senegal, Burkina-Faso, Ivory Coast, and Mali; and GFAOP 7 includes GFAOP 6 + Togo.
—, not applicable; CHOP-like, cyclophosphamide, doxorubicin, vincristine, and prednisone; COPM, cyclophosphamide, vincristine, prednisone, and hdMTX for 2 cycles, followed by hdMTX plus mini-cytarabine (500 mg/m2 per cycle) for 2 cycles; cyclophos, cyclophosphamide monotherapy; Doxo, doxorubicin; EFS, event-free survival; hdMTX, high-dose MTX; NR, not reported; Pred, prednisone; TRM, treatment-related mortality; VCR, vincristine.
The various regimens used in Uganda from 1967 to 1977 included cyclophosphamide monotherapy (1200 mg/m2 per cycle), COM (cyclophosphamide 900 mg/m2 per cycle, vincristine 2 mg/m2 per cycle, and MTX 45 mg/m2 per cycle), and cyclophosphamide 1200 mg/m2 per cycle, vincristine 1.4 mg/m2 per cycle, MTX 60 mg/m2 per cycle, and cytarabine, 750 mg/m2 per cycle.
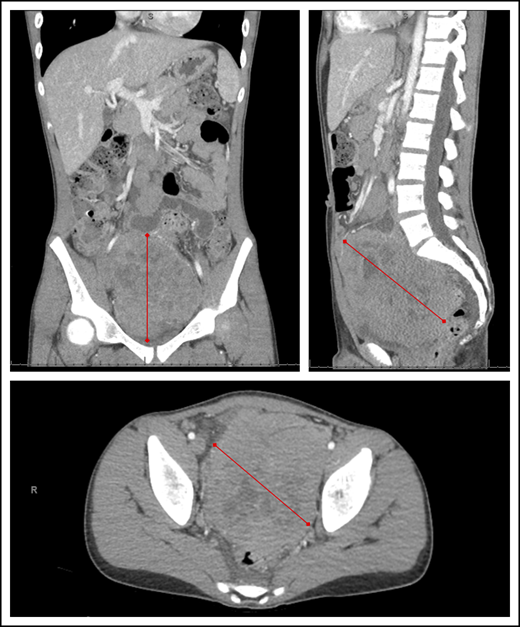
Abdominal computed tomography imaging demonstrating a large heterogeneous mass centered within the pelvis and lower abdomen in a child with BL. Red lines represent the extent of the lymphoma mass from its outer edges.
Abdominal computed tomography imaging demonstrating a large heterogeneous mass centered within the pelvis and lower abdomen in a child with BL. Red lines represent the extent of the lymphoma mass from its outer edges.
A recent clinical quality improvement project at the Uganda Cancer Institute is emblematic of the lack of progress in improving outcomes: over a 5-year period (2012-2017), 180 children with pathology-confirmed diagnoses of BL were treated with COM, with resources dedicated to ensuring accurate diagnosis, chemotherapy availability, and treatment compliance.44 Despite mitigating limitations of access to care, the 4-year OS was 44%,44 similar to results from clinical trials at the same center from the 1970s.53 Although the International Network for Cancer Treatment and Research had earlier reported event-free survival (EFS) and OS of 54% and 67%, respectively, at 1 year using a similar regimen with a slightly higher dose of MTX (75 mg/m2 compared with doses of 15-45 mg/m2 used in other COM trials), that study built in a salvage regimen of ifosfamide, etoposide, and cytarabine for patients who failed COM. Thus, the survival rates are not reflective of the treatment response to COM alone. Follow-up time for survivors was not reported for the cohort, creating uncertainty about whether the reported outcomes represented short-term response vs long-term remission.43 These survival rates have not been reproduced elsewhere in the region using similar regimens.
Another contemporary therapeutic strategy to improve on the suboptimal results of historic cyclophosphamide-based regimens has been based on the CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) adult mature B-cell NHL regimen. A study from Blantyre, Malawi reported a 1-year disease-free survival of 66% for patients with stage III/IV BL utilizing a regimen that added 2 doses of doxorubicin to a standard backbone of cyclophosphamide, vincristine, and prednisone.54 However, a similar regimen implemented in Nairobi, Kenya reported a 1-year EFS of 31%, whereas CHOP-like regimens used in Lilongwe, Malawi resulted in an 18-month OS of 29% in 2 cohorts (1 retrospective and 1 prospective).17,46,47 Notably, the disparate outcomes from the studies reported from Blantyre and Lilongwe, Malawi were not due to treatment-related mortality (TRM), which were very similar (8%-16%). It is challenging to definitively explain the differences in treatment outcomes, but potential factors may include differences in inclusion criteria, methods of risk stratification, response measurement, or follow-up.55 Additionally, the cohort in Blantyre may have benefited from expedited pathology analyses through the aforementioned telepathology collaboration via a partnership with a United Kingdom–based institution, potentially enabling a patient population with less-advanced-stage disease.21 Ultimately though, the less favorable outcomes from the region are similar to historic results from St. Jude Children’s Research Hospital (Memphis, TN) from the 1970s, where a similar regimen combining cyclophosphamide, vincristine, prednisone, and doxorubicin, with or without involved-field radiation therapy, resulted in a 2-year disease-free survival of 38.8% for patients with stage III/IV disease.56
In summary, despite the promising glimpse of responses and occasional cure for children with endemic BL receiving treatment in SSA in the 1970s, progress has stalled over the past 50 years, with low-intensity cyclophosphamide-based chemotherapy as a persistent therapeutic strategy.20 Although studies are largely retrospective with variable data integrity, at best, this chemotherapy approach affords long-term survival to approximately a third of children.
Role of high-dose MTX in pediatric BL regimens in HICs
In the 1970s and 1980s, chemotherapy regimens and treatment outcomes for children in SSA were similar to their counterparts in the United States.57-60 For example, 2-year EFS at St. Jude Children’s Research Hospital was 37% for patients treated prior to 1975. In the 1980s, the US collaborative Children’s Cancer Group studied a regimen consisting of cyclophosphamide, vincristine, MTX (at a dose of 300 mg/m2, higher than that typically used in SSA but logarithmically lower than contemporary HIC dosing), and prednisone, with or without daunorubicin. The addition of an anthracycline in this trial did not influence 10-year EFS, which remained modest at 55% (without daunorubicin) vs 57% (with daunorubicin).60 In stark contrast to SSA, treatment protocols for BL in the United States and Europe iteratively evolved through the 1980s and 1990s to intensive multiagent regimens with a risk-adapted approach, achieving long-term survival rates that now surpass 90%.16,58,59,61,62
The 2 most widely used protocols for pediatric BL worldwide are derived from the French-American-British (FAB)/Lymphomes Malins B (LMB) collaboration and the Berlin-Frankfurt-Münster (BFM) consortium; both groups use similar risk-stratification systems, with stage III/IV disease and elevated lactate dehydrogenase (LDH) levels serving as determinants of the need to intensify therapy (Table 2).9-12,15,61,63,64 In both constructs, the lowest-risk groups of patients (group A and group R1) are those with completely resected stage I or II tumors. This subgroup of patients is very rare in SSA. Therefore, the vast majority of patients with endemic BL are categorized as being in the analogous intermediate-risk R2/R3 and group B subcategories, as well as in the highest-risk R4/group C subsets.
Differential dosing of high-dose MTX based on risk stratification
| BFM risk group | MTX dose | Infusion time, h | FAB/LMB risk group | MTX dose | Infusion time, h |
|---|---|---|---|---|---|
| R1: stage I/II resected | 1000 mg/m2 × 2 | 4 | Group A: stage I/II resected | None | None |
| R2: unresected stage I/II, stage III with low LDH | 1000 mg/m2 × 4 | 4 | Group B low risk: not in groups A or C, low LDH | 3000 mg/m2 × 4 | 3 |
| R3: stage III and CNS− stage IV with intermediate LDH | 5000 mg/m2 × 4* | 24 | Group B high risk: not in groups A or C, high LDH | 3000 mg/m2 × 4 | 3 |
| R4: stage III/IV with high LDH or any CNS+ stage IV | 5000 mg/m2 × 4* | 24 | Group C: bone marrow ≥ 25% and/or CNS+ | 8000 mg/m2 × 3* | 4 for CSF−and 24 for CSF+ |
| BFM risk group | MTX dose | Infusion time, h | FAB/LMB risk group | MTX dose | Infusion time, h |
|---|---|---|---|---|---|
| R1: stage I/II resected | 1000 mg/m2 × 2 | 4 | Group A: stage I/II resected | None | None |
| R2: unresected stage I/II, stage III with low LDH | 1000 mg/m2 × 4 | 4 | Group B low risk: not in groups A or C, low LDH | 3000 mg/m2 × 4 | 3 |
| R3: stage III and CNS− stage IV with intermediate LDH | 5000 mg/m2 × 4* | 24 | Group B high risk: not in groups A or C, high LDH | 3000 mg/m2 × 4 | 3 |
| R4: stage III/IV with high LDH or any CNS+ stage IV | 5000 mg/m2 × 4* | 24 | Group C: bone marrow ≥ 25% and/or CNS+ | 8000 mg/m2 × 3* | 4 for CSF−and 24 for CSF+ |
CSF+ is defined by the presence of blasts on CSF cytology. LDH thresholds for BFM: low is <2 times the upper limit of normal (ULN), intermediate is 2 to 4 times the ULN, high is >4 times the ULN. LDH thresholds for FAB/LMB: low LDH is <2 times the ULN, and high LDH is >2 times the ULN.
Patients in risk groups R3, R4, and C also received high-dose cytarabine.
In the 1980s and 1990s, the French LMB and German BFM collaborative groups, along with St. Jude Children’s Research Hospital, pioneered the use of short intensive combination chemotherapy protocols, including alkylating agents, high-dose MTX (hdMTX; for intermediate and high-risk patients), and high-dose cytarabine (for high-risk patients), to achieve long-term survival in the majority of children with BL.62,63,65-67 This approach has optimized the systemic and CNS benefits of hdMTX and cytarabine and established the importance of intensity (ie, fewer shorter higher-dose chemotherapy cycles) compared with the prolonged, but lower-intensity, treatment regimens of the past.59 This has become the universal standard of treatment of BL.15,16,68,69 Despite the high intensity of cytotoxic chemotherapy used in these regimens, intrathecal chemotherapy remains an essential component of the standard BL regimen to treat or prevent CNS disease.61,70,71
The strongest evidence for the indispensable role of hdMTX in improving curative outcomes in children with advanced-stage BL stems from the work of Reiter et al in their 1999 publication comparing the BFM86 and BFM90 protocols.62 The 2 regimens were essentially identical with the exception of 1 major adjustment: an increase in MTX dose from 500 mg/m2 (BFM86) to 5000 mg/m2 (BFM90).62,72 The impact of this MTX dose intensification, which at the time was administered as a 24-hour infusion for all patients, was greatest in patients with abdominal stage III disease and LDH ≥500 U/L. The 6-year EFS for this subset of patients with advanced-stage disease increased from 43% in BFM86 to 81% in BFM90.62 These data highlight the critical importance of hdMTX in curative therapy for pediatric BL.
Although there are slight differences between the LMB and BFM regimens, they have a similar structure. The chemotherapy agents consist of hdMTX, alkylating agents (cyclophosphamide only in LMB, and alternating cyclophosphamide/ifosfamide in BFM), doxorubicin, vincristine, cytarabine, etoposide, and glucocorticoids (Tables 3 and 4). Recent reviews of the treatment of pediatric BL/diffuse large B-cell lymphoma (DLBCL) have extensively described and compared the details of these regimens.61,70,71 The use of high-intensity chemotherapy and immune-depleting agents comes with significant risk for end-organ damage and TRM. High-dose cytarabine and the combination of doxorubicin with MTX are associated with significant infectious morbidity.11 High doses of MTX are also associated with risk for acute kidney injury and severe mucositis. Pediatric cancer treatment programs in HICs have successfully mitigated the severity and occurrence of these adverse events with comprehensive supportive care measures and routine use of supportive pharmacological agents, such as folinic acid, hyperhydration with bicarbonate-based IV fluids, granulocyte colony-stimulating factor, empiric broad-spectrum antibacterial treatment in the setting of febrile illnesses, and blood product support. More recently, even for the highest-risk patients, the addition of rituximab (an anti-CD20 monoclonal antibody) to the standard chemotherapy backbones has propelled curative outcomes beyond 90%.13,14,16,73
Schematic comparison of the FAB/LMB and BFM risk-stratified chemotherapy regimens
| Risk strata | Regimen | Prephase | Chemotherapy phases | |||||
|---|---|---|---|---|---|---|---|---|
| Low risk | BFM R1 | None | A | B | ||||
| FAB/LMB group A | None | COPAD | COPAD | |||||
| Intermediate risk | BFM R2 | V | A | B | A | B | ||
| FAB/LMB group B | COP | COPADM | COPADM | CYM | CYM | |||
| BFM R3 | V | AA | BB | CC | AA | BB | ||
| High risk | FAB/LMB group C | COP | COPADM | COPADM2 | CYVE | CYVE | m1 | m2 |
| BFM R4 | V | AA | BB | CC | AA | BB | CC | |
| Risk strata | Regimen | Prephase | Chemotherapy phases | |||||
|---|---|---|---|---|---|---|---|---|
| Low risk | BFM R1 | None | A | B | ||||
| FAB/LMB group A | None | COPAD | COPAD | |||||
| Intermediate risk | BFM R2 | V | A | B | A | B | ||
| FAB/LMB group B | COP | COPADM | COPADM | CYM | CYM | |||
| BFM R3 | V | AA | BB | CC | AA | BB | ||
| High risk | FAB/LMB group C | COP | COPADM | COPADM2 | CYVE | CYVE | m1 | m2 |
| BFM R4 | V | AA | BB | CC | AA | BB | CC | |
A, cycle A; AA, cycle AA; B, cycle B; BB, cycle BB; CC, cycle CC; COP, COP pre-phase; COPADM, induction phase; COPADM2, induction phase; CYM, consolidation phase; CYVE, consolidation phase; m1, maintenance phase 1; m2, maintenance 2; V, pre-phase.
Comparison of chemotherapy combinations and doses in the FAB/LMB and BFM regimens
| Regimen | Phase of therapy | Chemotherapeutic agents and doses per phase, mg/m2 | IT doses, n | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cycl | VCR | P or D | Doxo | MTX | Cyta | Etop | Ifos | |||
| BFM | V | 400 | — | D: 40 | — | — | — | — | — | 1 |
| A | — | 1.5* | D: 50 | — | 1000 | 600 | 200 | 4000 | 1 | |
| B | 1000 | 1.5* | D: 50 | 50 | 1000 | — | — | — | 1 | |
| AA | — | 1.5 | D: 50 | — | 5000 | 600 | 200 | 4000 | 2† | |
| BB | 1000 | 1.5 | D: 50 | 50 | 5000 | — | — | — | 2† | |
| CC | — | 3* | D: 100 | — | — | 12 000 | 500 | — | 1† | |
| LMB | COP | 300 | 1 | P: 420 | — | — | — | — | — | 1† |
| COPAD | 1500 | 4 | P: 360 | 60 | — | — | — | — | 0 | |
| COPADM | 1500 | 2 | P: 300 | 60 | 3000-8000‡ | — | — | — | 2† | |
| COPADM2 | 3000 | 2 | P: 300 | 60 | 8000 | — | — | — | 3 | |
| CYM | — | — | — | — | 3000 | 500 | — | — | 2 | |
| CYVE | — | — | — | — | 8000§ | 12 250 | 800 | — | 1-2§ | |
| m1 | 1000 | 2 | P: 300 | 60 | 8000 | — | — | — | 1 | |
| m2 | — | — | — | — | — | 500 | 450 | — | 0 | |
| Regimen | Phase of therapy | Chemotherapeutic agents and doses per phase, mg/m2 | IT doses, n | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cycl | VCR | P or D | Doxo | MTX | Cyta | Etop | Ifos | |||
| BFM | V | 400 | — | D: 40 | — | — | — | — | — | 1 |
| A | — | 1.5* | D: 50 | — | 1000 | 600 | 200 | 4000 | 1 | |
| B | 1000 | 1.5* | D: 50 | 50 | 1000 | — | — | — | 1 | |
| AA | — | 1.5 | D: 50 | — | 5000 | 600 | 200 | 4000 | 2† | |
| BB | 1000 | 1.5 | D: 50 | 50 | 5000 | — | — | — | 2† | |
| CC | — | 3* | D: 100 | — | — | 12 000 | 500 | — | 1† | |
| LMB | COP | 300 | 1 | P: 420 | — | — | — | — | — | 1† |
| COPAD | 1500 | 4 | P: 360 | 60 | — | — | — | — | 0 | |
| COPADM | 1500 | 2 | P: 300 | 60 | 3000-8000‡ | — | — | — | 2† | |
| COPADM2 | 3000 | 2 | P: 300 | 60 | 8000 | — | — | — | 3 | |
| CYM | — | — | — | — | 3000 | 500 | — | — | 2 | |
| CYVE | — | — | — | — | 8000§ | 12 250 | 800 | — | 1-2§ | |
| m1 | 1000 | 2 | P: 300 | 60 | 8000 | — | — | — | 1 | |
| m2 | — | — | — | — | — | 500 | 450 | — | 0 | |
Cycl, cyclophosphamide; Cyta, cytarabine; D, dexamethasone; Etop, etoposide; Ifos, ifosfamide; IT, intrathecal; P, prednisone.
Vincristine is omitted from the BFM R1 regimen and is replaced by vindesine in the CC phase of therapy.
Additional IT therapy is administered to patients with CNS involvement in the BFM regimen, as well as for patients in LMB group C.
Dose of MTX is 3000 mg/m2 for patients in group B and 8000 mg/m2 for patients in group C in LMB.
High-dose MTX and an additional dose of intrathecal chemotherapy is given to patients with CNS involvement during 1st CYVE phase only.
Another important nuance in the difference between the BFM and LMB regimens arises in the duration of infusion time of hdMTX (Table 2). The early BFM regimens uniformly administered hdMTX over 24 hours.62,72 The BFM95 study compared 4- vs 24-hour MTX infusions and demonstrated that regimens with 4-hour infusions were significantly less toxic, with a lower incidence of grade III/IV mucositis. However, although treatment with 4-hour infusions was not inferior to 24-hour infusions for patients with lower-risk R1 and R2 disease, the probability of failure-free survival at 1 year was inferior for risk groups R3 and R4 (77% for those receiving 4-hour infusions compared with 93% for those receiving 24-hour infusions; P = .0077).63 Based on these data, BFM regimens stratify the intensity of MTX dosing based on risk group, adjusting the dose and infusion time (Table 2). The LMB regimen also adjusts the infusion time of hdMTX, reserving the 24-hour infusion duration for CNS+ patients who had blasts identified in the CSF. Otherwise, intermediate-risk group B patients receive MTX over a 3-hour duration and CSF− group C patients receive it over 4 hours, with the most recent international clinical trial demonstrating a 3-year EFS of 93.9% with rituximab plus LMB backbone chemotherapy for the high-risk group B and group C patients (Table 2).16 Thus, although the BFM95 data demonstrated superior outcomes for advanced-stage patients receiving the more intense 24-hour MTX infusions, the favorable outcomes for CSF− patients receiving shorter infusions in the LMB experience suggest that 24-hour infusions may be reserved for CSF+ patients only. One must also consider the increased toxicity associated with longer-duration MTX infusions, particularly in settings with limited resources for supportive care, where severe mucositis and/or acute kidney injury may have drastic consequences.
The use of current LMB- or BFM-based treatment regimens is limited to tertiary-level pediatric centers that have infrastructure for essential pathology services, diagnostic imaging and laboratory infrastructure for accurate risk stratification, and advanced capacity for supportive care, including intensive care units, blood banking services, and adequate nutritional support. As a result, these advances have mostly benefitted children in HICs and have not been adopted in low- and middle-income countries (LMICs), including SSA, where the vast majority of children with BL live, underlining the wide disparity in survival of children with BL.
Advancing treatment of BL in SSA
Improving diagnosis and risk stratification of endemic BL
Establishing an accurate diagnosis is a critical first step to improving the survival of children with BL in SSA (Figure 2). The most common limitation in the diagnostic accuracy of BL in LMICs is the reliance on clinical diagnosis in children with jaw tumors and morphology alone when tumor tissue is obtained, typically by fine needle aspiration.17 Our experience in SSA is that abdominal tumors are as common as jaw tumors as a presenting feature of BL in children, creating a diagnostic conundrum in differentiating between other abdominal lymphoma diagnoses, as well as the common solid tumors of the abdomen (ie, Wilms tumor, neuroblastoma, and germ cell tumors).17 To further complicate matters, rhabdomyosarcoma and lymphoblastic lymphoma may also present as jaw masses. Advanced orbital retinoblastoma can be indistinguishable from a maxillary or periorbital BL mass. Furthermore, the morphologic appearance of small round blue cells is not unique to BL; it is typical of other common pediatric malignancies, including lymphoblastic lymphoma, rhabdomyosarcoma, Wilms tumor, Ewing sarcoma, retinoblastoma, and neuroblastoma, among others. Even nonmalignant lymphadenopathy has been mistakenly diagnosed as BL.17-19 Alas, treatment outcomes are only as reliable as the diagnostic accuracy of the cohort of patients. Although cytoplasmic vacuolations, tinged body macrophages, and starry sky appearance are all morphological features considered unique to BL, they can be absent, and their diagnostic specificity is unknown; however, studies of diagnostic accuracy of morphology alone suggest that the specificity is ∼50% at best.18,19
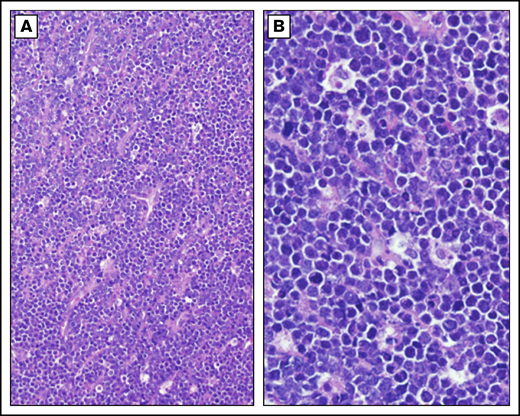
Burkitt lymphoma histopathology. Representative histological images from a child with BL demonstrating tumor morphology on hematoxylin and eosin–stained biopsy at low-power (A) and high-power (B) magnification.
Burkitt lymphoma histopathology. Representative histological images from a child with BL demonstrating tumor morphology on hematoxylin and eosin–stained biopsy at low-power (A) and high-power (B) magnification.
Establishment of laboratory infrastructure to ensure access to the World Health Organization minimum criteria, including immunophenotyping by immunohistochemistry or multiparametric flow cytometry for diagnosis of BL, is feasible in most countries in SSA. Flow cytometry is particularly advantageous because it can use tumor samples obtained by fine needle aspiration and precludes the need for general anesthesia, enables concurrent diagnoses of other hematological malignancies, is timelier than immunohistochemistry, and can be built on existing HIV immunology research infrastructure that is now widely available in many countries in SSA.17,22,23,74-79
The current standard for treating pediatric BL in HICs requires an accurate diagnosis to distinguish NHL arising from germinal center B cells (ie, BL and DLBCL), which are treated the same, vs other NHLs, such as those arising from T cells or precursor B cells, which require a different therapeutic strategy. In addition to morphological features consistent with lymphoid malignant proliferation, accurate diagnosis of BL requires immunophenotyping to demonstrate B-cell lineage of the malignant cells (eg, CD20, CD10) and to demonstrate the maturity of the malignant clone (eg, terminal deoxynucleotidyl transferase–negative and κ or λ surface immunoglobulin restriction). Although pediatric BL and DLBCL are treated the same in current standardized protocols, the 2 can sometimes be distinguished by a very high proliferative index in the case of BL, using Ki67, and demonstration of translocations involving C-MYC and any of the 3 immunoglobulin genes (IGH, IGΚ, or IGL). Risk-adapted treatment requires accurate risk stratification to tailor chemotherapy intensity to risk of relapse. Comprehensive staging evaluation requires light microscopy of a CSF cytospin sample and bone marrow aspirate or biopsy, in addition to computed tomography diagnostic imaging. The standard risk-stratification platform incorporates the pediatric NHL St. Jude Research Hospital’s staging classification and baseline LDH level.9-11,15,63 Technology for accurate staging and risk stratification are limited but are gradually becoming available at major medical academic centers in SSA and should continue to be prioritized. In addition, the development of telepathology infrastructure through global twinning partnerships can play an important role in scaling up local pathology expertise in the long term, as well as enhancing the accuracy and efficiency of diagnostic pathology in the short term.21-23
Escalating MTX dosing tailored to available supportive care in SSA
Current best evidence is that high-intensity cytotoxic chemotherapy is critical to cure BL, particularly in children with intermediate- and high-risk disease. Based on experiences with the LMB and BFM regimens discussed above, MTX is a critical agent in the cure of BL. Given the inability of pediatric centers in SSA to provide the level of supportive care necessary to safely implement the full complement of current gold standard LMB- and BFM-based regimens, graduated and step-wise escalation of MTX doses should be tailored to the available supportive care infrastructure. Although concerted efforts continue to focus on improving the clinical infrastructure in centers in SSA, this should not negate attempts to advance beyond low-dose MTX and low-intensity cyclophosphamide regimens that have stalled progress in improving survival of African children with BL in the past 50 years.
In Blantyre, Malawi, a regimen using 2000 mg/m2 MTX (infused over 3 hours), in combination with cyclophosphamide and vincristine, resulted in a disappointing 1-year EFS of 33%, with a high TRM of 33%; any potential gains made by intensification of MTX were probably lost in TRM.80 Notably, this regimen used a dose of cyclophosphamide (500 mg/m2) that is lower than typically used, even in SSA regimens.
Around the same time period, from 2001 to 2004, the Groupe Franco-Africain d’Oncologie Pédiatrique (GFAOP) collaboration in Cameroon, Madagascar, and Senegal escalated pediatric BL treatment from cyclophosphamide monotherapy (1-year EFS = 33%) to cyclophosphamide, vincristine, MTX, and prednisone, using an MTX dose of 3000 mg/m2 (infused over 3 hours).41 This higher-intensity regimen was initially characterized by high TRM rates (26% in the first year) at GFAOP centers; however, with ongoing improvements in supportive care, TRM decreased with each year of the study (second year TRM = 19%, third year TRM = 12%).81 The authors attributed these improvements to enhanced prevention and treatment of infections and tumor lysis syndrome.81
More recently, the GFAOP published a larger (n = 400) multicenter experience involving 7 sites in SSA. They were able to reproduce the superior outcomes seen in the earlier experience with a 1-year OS of 60% for patients with advanced-stage disease and a TRM of 12%.51 Using a modified LMB regimen, they continued to deliver the first 2 induction cycles with cyclophosphamide, vincristine, prednisone, and hdMTX for 2 cycles, followed by hdMTX plus mini-cytarabine (500 mg/m2 per cycle) for 2 cycles, including 3000 mg/m2 of MTX over 3 hours.51 The progress made by GFAOP highlights the necessity for enhanced supportive care along with intensified MTX.
How much MTX is enough?
Doses and infusion duration of MTX used in the treatment of pediatric BL in the United States and Europe have varied over time and depend on risk stratification (Table 2). The BFM86 protocol, which administered 500 mg/m2 of MTX over 24 hours, reported a 6-year EFS of 99% for patients with stage I/II disease.62,72 In BFM95, patients in low-intermediate–risk groups R1 and R2 received 1000 mg/m2 over 4 hours and achieved a similar 3-year EFS of 98%; this dose has been used for R1 and R2 strata in the BFM regimen ever since.63 For higher-risk R3 and R4 patients, higher doses of MTX with longer infusions times were considered necessary. As previously mentioned, the BFM90 regimen used 5000 mg/m2 of MTX over 24 hours to achieve improved long-term EFS in patients with advanced-stage disease (Table 5).62
Outcomes for childhood and adolescent mature B-cell NHL with historical treatment regimens from Europe plus middle-income countries
| Location | Years | Sample size, n | Regimen basis | MTX dose utilized, mg/m2 | Survival analysis | Cohort survival, % | TRM, % | Survival by stage/risk group |
|---|---|---|---|---|---|---|---|---|
| BFM | 1986-1990 | 197 | BFM86 | 500 | 6-y EFS | 82 | 5 | Stages I/II, 99%; stage III LDH < 500 U/L, 85%; stage III LDH > 500 U/L, 43%; stage IV, 67% |
| BFM | 1990-1995 | 413 | BFM90 | 5000 | 6-y EFS | 89 | 3 | Stages I/II, 98%; stage III LDH < 500 U/L, 95%; stage III LDH > 500 U/L, 81%; stage IV, 73% |
| BFM | 1996-2001 | 281 | BFM95* | 1000 | 3-y EFS | 94 | 0.4 | Group R1, 94%; group R2, 94% |
| Brazil | 1998-2003 | 53 | BFM86/90 | 2000 | ∼3-y EFS | 78 | 2 | Stages I/II, 100%; stages III/IV, 74% |
| Argentina | 1994-1999 | 57 | BFM90 | 2000 | 5-y EFS | 79 | 9 | Group R1, 100%; group R2, 86%; group R3, 82%; group R4, 50% |
| Nicaragua | 1996-2003 | 53 | Unspecified | 1000 | 9-y EFS | 53 | 2 | Stage I, 100%; stage II, 71%; stage III, 55%; stage IV, 17% |
| Venezuela | 1995-2002 | 96 | LMB89 | Unmodified | 2-y EFS | 75 | 9 | Group A, 100%; group B, 76%; group C, 56% |
| South Africa | 1995-2010 | 51 | LMB89 | Unmodified | 5-y OS | 65 | NR | Stage II, 62%; stage III, 78%; stage IV, 42% |
| Northwest Africa | 2001-2004 | 119 | LMB89 | Unmodified | 3-y OS | 75 | 21 | Stages I/II, 84%; stage III, 76%; stage IV, 56% |
| Central America | 2004-2016 | 405 | BFM90 | 1000-3000 | 3-y EFS | 70 | 11 | Stages I/II, 83%; stage III, 75%; stage IV, 55% |
| Location | Years | Sample size, n | Regimen basis | MTX dose utilized, mg/m2 | Survival analysis | Cohort survival, % | TRM, % | Survival by stage/risk group |
|---|---|---|---|---|---|---|---|---|
| BFM | 1986-1990 | 197 | BFM86 | 500 | 6-y EFS | 82 | 5 | Stages I/II, 99%; stage III LDH < 500 U/L, 85%; stage III LDH > 500 U/L, 43%; stage IV, 67% |
| BFM | 1990-1995 | 413 | BFM90 | 5000 | 6-y EFS | 89 | 3 | Stages I/II, 98%; stage III LDH < 500 U/L, 95%; stage III LDH > 500 U/L, 81%; stage IV, 73% |
| BFM | 1996-2001 | 281 | BFM95* | 1000 | 3-y EFS | 94 | 0.4 | Group R1, 94%; group R2, 94% |
| Brazil | 1998-2003 | 53 | BFM86/90 | 2000 | ∼3-y EFS | 78 | 2 | Stages I/II, 100%; stages III/IV, 74% |
| Argentina | 1994-1999 | 57 | BFM90 | 2000 | 5-y EFS | 79 | 9 | Group R1, 100%; group R2, 86%; group R3, 82%; group R4, 50% |
| Nicaragua | 1996-2003 | 53 | Unspecified | 1000 | 9-y EFS | 53 | 2 | Stage I, 100%; stage II, 71%; stage III, 55%; stage IV, 17% |
| Venezuela | 1995-2002 | 96 | LMB89 | Unmodified | 2-y EFS | 75 | 9 | Group A, 100%; group B, 76%; group C, 56% |
| South Africa | 1995-2010 | 51 | LMB89 | Unmodified | 5-y OS | 65 | NR | Stage II, 62%; stage III, 78%; stage IV, 42% |
| Northwest Africa | 2001-2004 | 119 | LMB89 | Unmodified | 3-y OS | 75 | 21 | Stages I/II, 84%; stage III, 76%; stage IV, 56% |
| Central America | 2004-2016 | 405 | BFM90 | 1000-3000 | 3-y EFS | 70 | 11 | Stages I/II, 83%; stage III, 75%; stage IV, 55% |
BFM95 data focus on regimen for risk groups R1 and R2 only, excluding risk groups R3 and R4, which received a higher dose of MTX.
In the current BFM regimen NCT03206671, patients in low-intermediate–risk groups R1 and R2 receive 1000 mg/m2 of MTX infused over 4 hours, whereas the intermediate-high–risk groups R3 and R4 receive 5000 mg/m2 infused over 24 hours.62,63,72 In the LMB regimens, intermediate-risk patients (group B) receive 3000 mg/m2 over 3 hours, whereas high-risk patients (group C) receive 8000 mg/m2 over 4 hours (CSF−) or over 24 hours (CSF+) (Table 2). An important caveat is that the LMB regimen incorporates 2 cycles containing high-dose cytarabine for group C patients, whereas the BFM regimen includes 1 cycle of high-dose cytarabine for risk group R3 and 2 cycles for risk group R4. The use of high-dose cytarabine is associated with a high risk for infectious morbidity and mortality due to prolonged severe neutropenia.11 Therefore, this subset of high-risk patients may present the greatest challenge to improving curative outcomes in low-income settings in SSA.
Nonetheless, other pediatric oncology centers in LMICs around the world have established precedent for improving curative outcomes for children with BL by delivering risk-stratified treatment based on adapted regimens, with or without dose modifications of hdMTX. Table 4 summarizes the nuances of intermediate-high–dose MTX (starting from 500 mg/m2) that have been used in pediatric BL.62,63,72,81-87 Multiple cohorts from Central and South America had long-term EFS rates ranging from 55% to 82% for patients with stage III BL utilizing regimens with intermediate-dose MTX (1000-2000 mg/m2), typically infused over 3 hours; the only exception was the study in Argentina, which was based on the BFM90 approach with 24-hour infusions.82-84,87 Although considerations to reduce MTX doses in HICs are counteracted by the dismal survival of children with relapsed BL, only lower doses of MTX are safely feasible in most SSA centers. It will be critical to determine the safest most effective dosing range and infusion duration to optimize curative outcomes in a risk-stratified manner for children with BL in low-income settings.
Potential impact of rituximab in the treatment of BL in SSA
The anti-CD20 monoclonal antibody rituximab has been incorporated into the treatment of pediatric BL for nearly 2 decades. It was initially used with combination chemotherapy in the setting of relapsed and refractory disease and has since been evaluated as upfront therapy for BL in multiple clinical trials over the past 15 years.13,14,64,88 Recently, an international randomized phase 3 trial comparing standard LMB chemotherapy with/without rituximab demonstrated superior 3-year EFS for high-risk group B and group C patients receiving rituximab (93.9% vs 82.3%).16 The study demonstrated an association with a higher incidence of hypogammaglobulinemia in patients receiving rituximab, as well as a trend toward a higher incidence of febrile neutropenia and infection; however, overall, the regimen was well tolerated.16 In light of the favorable impact of rituximab on children with mature B-cell NHL, clinical trials in Europe (NCT03206671) and the United States (NCT01859819) have been pursuing the potential to reduce the burden of the BFM and LMB regimens through omission or dose reduction of doxorubicin in lower-risk patients receiving rituximab.73 Results from these therapy-reduction trials will have potential implications for the treatment of BL in SSA; if the addition of rituximab enables a decrease in the intensity of traditional cytotoxic chemotherapy, this would emphasize the importance of increasing access to rituximab for children with BL and DLBCL living in LMICs. The feasibility of giving rituximab to adults with lymphoma in Africa has already been established, and its incorporation in the treatment of pediatric mature B-cell NHL in Eastern Europe has demonstrated efficacy, despite reducing the intensity of induction chemotherapy.89,90 Ideally, the addition of rituximab to chemotherapy regimens for children with BL in Africa will enable the establishment of safer less-intense multiagent chemotherapy regimens that can optimize treatment response while mitigating the severe consequences of treatment toxicity.
Next steps in optimizing hdMTX in SSA
Optimization of MTX dose and infusion duration at treatment centers in SSA will vary based on locally available resources, including the multidisciplinary support required to safely and effectively administer the drug. Practical issues, such as night-staff availability and access to resources required for hyperhydration and leucovorin rescue, are critically important. One consideration is to escalate MTX dosing in a stepwise fashion, starting with a modest dose, such as 1000 mg/m2 infused over 3 hours. This dose has demonstrated efficacy for patients categorized as R2 in the BFM regimen and would at least include those with stage I/II disease, as well as stage III with low LDH. As centers establish safety with this initial dose, stepwise escalation up to 3000 mg/m2 infused over 3 hours will be a reasonable goal. This dose has proven efficacy for high-risk group B patients in the LMB regimen, including those with stage III disease and elevated LDH, extending curative potential to the majority of patients with BL. Escalation beyond these doses, and to longer infusions of MTX, may be goals reserved for future time points when treatment centers have greater infrastructure to deliver such intensive regimens.
Conclusions
Pediatric BL is emblematic of the disparities in survival of children with cancer worldwide. Advances in pediatric oncology in the United States and Europe blossomed through the development of clinical trial networks. In Africa, collaborative programs turned the tides of the HIV/AIDS epidemic.91 Collaborative clinical trial networks like the GFAOP are now required to systematically catalyze outcomes for children with cancer in SSA.91 Multidisciplinary specialized services, including nursing, pharmacy, nutrition, diagnostic pathology, blood banking, radiology, palliative care, pediatric surgery, and critical care, are key to improving pediatric cancer survival. Rational and feasible interim measures to improve outcomes of children with cancer in these settings should focus on adapting current best evidence to the resources, infrastructure, and putative unique biology of some of the cancers in children in LMICs. In the case of BL, the GFAOP approach of escalating MTX with concomitant supportive care through multicenter clinical trials performed through a cooperative group structure has improved outcomes.51 Careful implementation of higher MTX doses with combination chemotherapy in the setting of systematic supportive care infrastructure building is prudent. This stepwise escalation with ongoing rigorous evaluation and measurement of TRM may also offer the opportunity to establish the correct balance of intensity of cytotoxic chemotherapy for BL in the context of wider accessibility to anti-CD20 humoral immunotherapy globally.89,90
Authorship
Contribution: N.W.O., J.L., and N.K.E.-M. designed the research, analyzed the data, and wrote the manuscript; and C.E.A. analyzed the data and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial conflicts.
Correspondence: Nader Kim El-Mallawany, Texas Children’s Cancer and Hematology Centers, Baylor College of Medicine, Global HOPE, Lymphoma and Histiocytosis Team, 1102 Bates St Feigin Tower, Room 1025.16, Houston, TX 77030; e-mail: nader.el-mallawany@bcm.edu.

