

Key Points
Circulating plasmodial histones are significantly elevated in CM and are associated with endothelial and clotting activation.
Parasite histones may contribute to the development of CM by triggering focal coagulation activation and endothelial injury in the brain.

Abstract
Microvascular thrombosis and blood–brain barrier (BBB) breakdown are key components of cerebral malaria (CM) pathogenesis in African children and are implicated in fatal brain swelling. How Plasmodium falciparum infection causes this endothelial disruption and why this occurs, particularly in the brain, is not fully understood. In this study, we have demonstrated that circulating extracellular histones, equally of host and parasite origin, are significantly elevated in CM patients. Higher histone levels are associated with brain swelling on magnetic resonance imaging. On postmortem brain sections of CM patients, we found that histones are colocalized with P falciparum–infected erythrocytes sequestered inside small blood vessels, suggesting that histones might be expelled locally during parasite schizont rupture. Histone staining on the luminal vascular surface colocalized with thrombosis and leakage, indicating a possible link between endothelial surface accumulation of histones and coagulation activation and BBB breakdown. Supporting this, patient sera or purified P falciparum histones caused disruption of barrier function and were toxic to cultured human brain endothelial cells, which were abrogated with antihistone antibody and nonanticoagulant heparin. Overall, our data support a role for histones of parasite and host origin in thrombosis, BBB breakdown, and brain swelling in CM, processes implicated in the causal pathway to death. Neutralizing histones with agents such as nonanticoagulant heparin warrant exploration to prevent brain swelling in the development or progression of CM and thereby to improve outcomes.
Introduction
Cerebral malaria (CM) is a severe complication of Plasmodium falciparum infection. Despite effective antimalarial drugs, 10% to 20% of children developing CM die,1 contributing to 400 000 malarial deaths per year, mostly in children in sub-Saharan Africa.2 Recent magnetic resonance imaging (MRI) studies demonstrated brain swelling in the causal pathway to death and implicated blood–brain barrier (BBB) breakdown.3,4 Death typically occurs in the first 24 hours after admission,5 with children who do not reach critical levels of brain swelling frequently recovering rapidly. BBB stabilization, through targeting causal pathways to vascular leak in the brain, could halt this brain swelling and reduce mortality.
A defining feature of CM is cytoadherence of P falciparum–infected erythrocytes (IE) to endothelial cells (EC) leading to sequestration in the microvasculature.1 In vivo retinal imaging,6,7 postmortem histology,8,9 and in vitro data10 demonstrate spatial-temporal links between sequestration, microvascular leak, and thrombosis, with coagulopathy predicting fatal outcome in CM.11,12 Postmortem studies in African children demonstrate sequestration in multiple organs, whereas leak and coagulopathy are most prominent in the brain9,13,14 ; this implies that sequestration provides a parasite stimulus for vascular leak and coagulopathy to which the brain is particularly vulnerable.8,15
Extracellular histones, released by damaged or immune-activated host cells have emerged as critical mediators of EC damage in diverse severe illnesses including sepsis,16 influenza,17 inflammatory conditions,18,19 and trauma.20 Hallmark features of histone toxicity are thrombocytopenia21,22 and microvascular thrombosis and leak.16,20 In patients with sepsis or trauma, histone levels correlate with clinical severity scores,20,23 thrombocytopenia,21 and coagulation activation,20,23,24 and they predict outcome.20 In animal models of sepsis or trauma, released histones are causal in these processes and in fatal outcome, prevented by antihistone antibodies20,23,25,26 and heparins27 (which neutralize histones), and by activated protein C (aPC), which degrades histones.16 In mice, infusion of exogenous histones of >30 mg/kg is toxic; infusions of >60 mg/kg are fatal. In histological sections, histones are observed to accumulate on the endothelial surface and colocalize with microvascular coagulopathy and vascular leak.20
The clinicopathological features observed at sites of sequestration in CM,8,9,14 vascular leak, coagulopathy, and thrombocytopenia, strongly resemble those that are induced by histones in other conditions.16,20 Therefore, we hypothesized that histones might be an important causal factor in CM pathogenesis. Both P falciparum and mammalian cells contain histones (H2A, H2B, H3, H4), packaged in nucleosomes with DNA. Following the sequestration of parasitized erythrocytes, intraerythrocytic merozoites multiply by up to 30 times to form a schizont. This increases the nuclear material, including histones, by an order of magnitude. When schizonts rupture, they release their contents; in vitro they can be shown to expel P falciparum histones.28,29 Purified plasmodial histones cause inflammatory pathway activation, toxicity, and barrier disruption on cultured endothelial cells, similar to mammalian histones.28 Therefore, histones may link sequestration and vascular pathology in CM. Histone-packed schizonts sequestered in red cells in contact with the endothelial surface may, upon rupture, deliver an intense, concentrated exposure to released histones, leading to coagulation activation and endothelial injury and, in turn, BBB breakdown and brain swelling.
It remains uncertain whether significant levels of parasite histones are produced in vivo in patients with malaria. There have been no data assessing the association between histones originating from host or parasite and clinical or laboratory indicators of endothelial or coagulation activation or disease severity in CM, nor have there been any data to assess whether histones accumulate on the vascular endothelium at sites of sequestration, thrombosis, or BBB breakdown. In this study, we address these gaps using samples from well-characterized CM patients, and, in ex vivo and in vitro experiments, demonstrate a causal role of P falciparum histones in endothelial disruption.
Methods
Children included in the study and patient and postmortem case definitions
Children aged 6 months to 16 years were recruited at Queen Elizabeth Central Hospital (Blantyre, Malawi) between January 2010 and August 2011 (Liverpool School of Tropical Medicine Research ethics protocol 09-74; Malawi College of Medicine Research Ethics Committee protocol number P.02/10/860). Inclusion criteria are described previously.8 Supplemental Figures 1 and 2 summarize which patients were included and excluded from the analysis. Children who met WHO criteria for CM (standard clinical definition of CM) underwent funduscopic examination by an ophthalmologist; characteristic retinal changes are associated with sequestration of IE in the brain.1,30,31 Children in coma but who did not have malaria (non-CM coma) were used as a comparator group. Children with uncomplicated malaria and mild nonmalarial febrile illness (mild febrile illness; malaria parasites not detected in blood on thick smear) were recruited from the hospital Accident and Emergency department. They had no evidence of organ compromise and were assessed to be well enough to go home. Healthy controls were children attending elective surgery.
Postmortem, cases who met WHO criteria for CM while alive were assessed by a clinical pathologist and divided into three groups: definitive CM cases, non-CM parasitemic cases, or nonmalaria coma cases. Definitive CM cases showed sequestration of IE in the brain and no other cause of death. Non-CM parasitemic cases showed no visible sequestration of IE in cerebral vessels and a nonmalarial cause of death identified. These cases are generally retinopathy negative ante-mortem. Nonmalaria coma cases, children who were admitted in coma and who did not have malaria (no parasites on thick smear), were used as a comparator group (postmortem cases detailed in supplemental Table 1).
Plasma and serum samples
Venous blood was collected at enrollment, and serum and plasma was prepared as previously described.32 Circulating histone levels were quantified in serum by a custom immunoblot assay.20,21,23 In plasma we quantified Osteoprotegerin (OPG; R&D Systems) and F1+2 peptide (Enzygnost; Siemens) by ELISA33,34 and fibrin monomers using an STA compact analyzer (Stago).11
Scoring of brain swelling
MRI images acquired on admission were scored independently by 2 radiologists blinded to patient details as described previously3 (for details see supplemental Methods). Briefly, patients were divided into 4 groups based on the scores: 1) no brain swelling, 2) mild brain swelling, 3) moderate brain swelling, and 4) severe brain swelling. A number of children did not have MRI scans. If a patient recovered from their coma within 12 hours and, thus, did not have an MRI scan, we deemed it likely that they did not have significant brain swelling and included them in the first group, no brain swelling. Other reasons for not obtaining MRI scans included clinical instability and equipment malfunction for whom we could not reasonably assign a category. Missing data were handled by listwise deletion.
Isolation, purification, and liquid chromatography–mass spectrometry analysis of histones
P falciparum histones (H2A/H2B, H3, and H4) were purified from saponin lysed IT4var16 IE using a Histone Purification Kit (Active Motif; detailed in supplemental Methods). Human histones (H1, H2A, H2B, H3, and H4) were purchased from New England Biolabs. Histones were enriched using gel separation, prior to liquid chromatography–mass spectrometry analysis. Analysis was performed using an Ultimate 3000 RSLC nano system (Thermo Scientific; Hemel Hempstead) coupled to a QExactive-Hf mass spectrometer (Thermo Scientific).
Immunohistochemistry
Formalin-fixed postmortem brain tissue samples from Malawian children with fatal encephalopathic illness were collected as described previously.9 Cortical sections (4 µm thick) were stained for histones and fibrinogen (supplemental Methods). Two scorers, blinded to histological classification, scored all of the cases, and a third scorer scored a subset of cases. The scorers used the same slide set, but after scanning the whole slide, selected a prespecified number of vessels at random from representative areas of the slide; 70 to 90 vessels were scored from each case. All scores were used for the analysis. IE sequestration for each vessel was scored as: negative (0); positive but <50% of the vessel lumen (+), or positive with >50% of the vessel lumen (++, high). Histone membrane staining for each vessel was scored as absent (0), weak (+), or strong (++). Fibrinogen extravasation as a marker of leak was scored for each vessel as absent or present.
Endothelial cell culture, endothelial cell damage assays, and barrier function assays
Primary human brain microvascular EC (HBMEC; Cell Systems) were cultured in 1% gelatin or fibronectin-coated (5 μg/mL) flasks, in complete medium (Cell systems) or endothelial growth medium 2 (Promocell, Germany) for transendothelial electrical resistance (TEER) experiments. For toxicity assays, confluent layers of HBMEC were treated with either purified histones or serum from healthy controls or patients (diluted 1:1 with serum-free media). Cell viability was determined by propidium iodide staining and quantified using flow cytometry.
Permeability of confluent HBMEC monolayers was analyzed in a dual-chamber system (0.4 µM pore size; Millipore, Germany), measuring horseradish peroxidase leak-through over 1 hour using tetramethylbenzidine substrate (supplemental Methods). TEER was measured using a real-time impedance system (xCELLigence; ACEA Biosciences; supplemental Methods).
For antihistone treatments, either purified histones or patient sera were preincubated for 10-15 minutes with antihistone single-chain variable fragment (ahscFv; 200 μg/mL20 ) or with nonanticoagulant N-acetyl heparin (200 μg/mL; Sigma).
Statistical analysis
Continuous variables were assumed to have normal or log normal distribution depending on their level of skewness. Differences between groups were compared using Student t tests for 2 group comparisons or analysis of variance (ANOVA) for multiple group comparisons. For ANOVA, to assess for differences between groups and adjust for multiple comparisons, we used the Tukey honestly significant difference test (when comparing all groups to each other) or Dunnett test (when comparing all groups to a single group). Differences between conditions for in vitro data were assessed using the Kruskal-Wallis test with a Dunn’s test to compare different samples to the control or the Friedman test to compare matched data to the control. The association between histone levels and other variables was assessed by linear regression and expressed as correlation coefficients (Pearson’s). For ordered categorical slide scoring data, the associations between histological classification, extent of sequestration, and degree of fibrinogen extravasation were assessed by use of ordinal logistic regression models.8 We adjusted for clustering among cases and scorers by including these as covariates in the regression model. All tests were 2-tailed with a conventional 5% significance level. Statistical analyses were performed using Stata (version 11; Statacorp) and Prism (version 8; GraphPad) software.
Results
Extracellular histones are elevated in CM
Clinical characteristics of the patients are detailed in Table 1. Circulating histone levels in patients with a clinical diagnosis of CM according to WHO criteria35 (geometric mean, 19.3 μg/mL; 95% confidence interval [CI], 15.4 to 24.3 μg/mL) were significantly higher than in healthy controls (geometric mean, 0.6 μg/mL; 95% CI, 0.2 to 1.7 μg/mL) and patients with mild febrile illness (geometric mean, 0.54 μg/mL; 95% CI, 0.3 to 1.4 μg/mL), uncomplicated malaria (geometric mean, 3.0 μg/mL; 95% CI, 2.0 to 4.5 μg/mL), and non-CM coma (geometric mean, 2.7 μg/mL; 95% CI, 0.6 to 12.0 μg/mL) (Figure 1A). We used retinal examination to refine CM diagnosis; characteristic retinal changes indicate sequestration of IE in the brain30 and distinguish retinopathy-positive CM (Ret+CM) cases from cases that are retinopathy-negative CM (Ret−CM) who are a more heterogeneous group and more likely to have an alternative diagnosis.1 Histone levels in Ret+CM (geometric mean, 24.7 μg/mL; 95% CI, 19.5 to 30.9 μg/mL) were markedly higher than those in Ret−CM cases (geometric mean, 8.3 μg/mL; 95% CI, 4.6 to 15.0 μg/mL; Figure 1B).
Clinical characteristics of the children
| Healthy controls (n = 22) | Mild febrile illness (n = 34) | UM (n = 50) | Nonmalarial coma (n = 10) | Ret−CM (n = 48) | Ret+CM (n = 170) | |
|---|---|---|---|---|---|---|
| Age, median (IQR), mo | 79 (43-107) | 41 (24-63) | 63 (40-92) | 46 (32-72) | 48 (28-69) | 47 (31-66) |
| Female sex, no. (%) | 9 (43) | 15 (44) | 26 (52) | 1 (10) | 23 (48) | 86 (51) |
| HIV positive, no. (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 4 (8.3) | 14 (8.2) |
| Axillary temperature, median (IQR), °C | 36.8 (36.1-36.8) | 38.2 (37.9-38.6) | 38.3 (37.9-39.0) | 38.6 (38.4-39.0) | 38.7 (37.7-39.6) | 38.7 (38.9-39.6) |
| Pulse rate, median (IQR), beats per minute | 116 (102-124) | 136 (113-154) | 137 (119-147) | 140 (119-157) | 143 (130-164) | 150 (138-167) |
| Systolic BP, median (IQR), mmHg | 111 (103-118) | 117 (107-123) | 114 (107-122) | 100 (94-110) | 98 (90-105) | 95 (89-104) |
| Respiratory rate, median (IQR), breaths/min | 28 (20-32) | 32 (28-36) | 27 (24-32) | 37 (28-40) | 40 (36-52) | 44 (38-52) |
| Blood glucose, median (IQR), mmol/L | 5.2 (4.6-5.7) | 4.8 (4.4-5.4) | 5.8 (4.9-6.4) | 7.45 (6.2-8.8) | 6.7 (5.5-8.6) | 6.4 (5.3-8.0) |
| Blood lactate, median (IQR), mmol/L | 1.8 (1.8-2.00) | 1.7 (1.2-2.2) | 2.4 (1.9-3.0) | 3.1 (2.1-5.2) | 4.0 (2.9-7.1) | 6.5 (3.6-10.3) |
| Hb, median (IQR), g/L | 103 (98-111) | 115 (105-120) | 93 (76-107) | 91 (82-92) | 81 (68-100) | 64 (51-77) |
| Platelets, median (IQR), ×109/L | 402 (345-470) | 331 (239-388) | 132 (82-185) | 335 (176-462) | 138 (60-221) | 52 (28-84) |
| Peripheral parasite density, median (IQR), ×103/μL | 0 | 0 | 31 (0.7-32) | 0 | 48 (5-173) | 73 (15-273) |
| Healthy controls (n = 22) | Mild febrile illness (n = 34) | UM (n = 50) | Nonmalarial coma (n = 10) | Ret−CM (n = 48) | Ret+CM (n = 170) | |
|---|---|---|---|---|---|---|
| Age, median (IQR), mo | 79 (43-107) | 41 (24-63) | 63 (40-92) | 46 (32-72) | 48 (28-69) | 47 (31-66) |
| Female sex, no. (%) | 9 (43) | 15 (44) | 26 (52) | 1 (10) | 23 (48) | 86 (51) |
| HIV positive, no. (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 4 (8.3) | 14 (8.2) |
| Axillary temperature, median (IQR), °C | 36.8 (36.1-36.8) | 38.2 (37.9-38.6) | 38.3 (37.9-39.0) | 38.6 (38.4-39.0) | 38.7 (37.7-39.6) | 38.7 (38.9-39.6) |
| Pulse rate, median (IQR), beats per minute | 116 (102-124) | 136 (113-154) | 137 (119-147) | 140 (119-157) | 143 (130-164) | 150 (138-167) |
| Systolic BP, median (IQR), mmHg | 111 (103-118) | 117 (107-123) | 114 (107-122) | 100 (94-110) | 98 (90-105) | 95 (89-104) |
| Respiratory rate, median (IQR), breaths/min | 28 (20-32) | 32 (28-36) | 27 (24-32) | 37 (28-40) | 40 (36-52) | 44 (38-52) |
| Blood glucose, median (IQR), mmol/L | 5.2 (4.6-5.7) | 4.8 (4.4-5.4) | 5.8 (4.9-6.4) | 7.45 (6.2-8.8) | 6.7 (5.5-8.6) | 6.4 (5.3-8.0) |
| Blood lactate, median (IQR), mmol/L | 1.8 (1.8-2.00) | 1.7 (1.2-2.2) | 2.4 (1.9-3.0) | 3.1 (2.1-5.2) | 4.0 (2.9-7.1) | 6.5 (3.6-10.3) |
| Hb, median (IQR), g/L | 103 (98-111) | 115 (105-120) | 93 (76-107) | 91 (82-92) | 81 (68-100) | 64 (51-77) |
| Platelets, median (IQR), ×109/L | 402 (345-470) | 331 (239-388) | 132 (82-185) | 335 (176-462) | 138 (60-221) | 52 (28-84) |
| Peripheral parasite density, median (IQR), ×103/μL | 0 | 0 | 31 (0.7-32) | 0 | 48 (5-173) | 73 (15-273) |
Hb, hemoglobin; IQR, interquartile range.
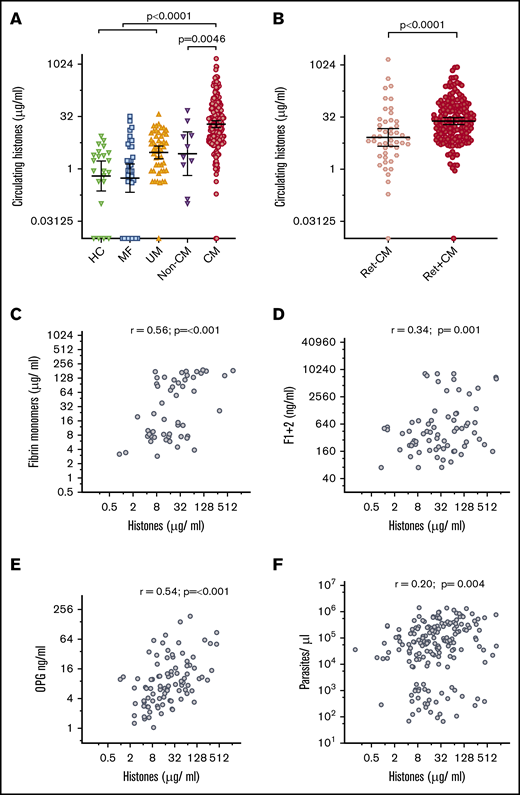
Circulating extracellular histones are elevated in CM. (A) Circulating histone levels measured in the serum of healthy controls (HC; n = 22), and patients with mild febrile illness (MF; n = 34), uncomplicated malaria (UM; n = 50), aparasitemic non-CM coma (Non-CM; n = 10) and admission serum from children who met the standard WHO case definition of CM at presentation (n = 218). Thick black lines and error bars are the geometric mean and 95% CI. (B) Refining the diagnosis of CM by retinal examination, comparison is made between circulating histone levels in children with Ret−CM (n = 48) and Ret+CM (n = 170). (C-F) Scatter plots to examine the correlation between histone levels and plasma factors in children with Ret+CM are shown. (C) Plasma fibrin monomer levels; (D) prothrombin fragment F1+2; (E) OPG; and (F) whole blood peripheral parasite density. (C-E) Data shown were measured by ELISA, and parasite density was calculated from white blood cell–normalized slide counts using thick smears. Comparison was made using ANOVA and Dunnett’s multiple comparison test (A), Student t test (B), and Pearson's correlation coefficient (C-F). Non-CM are aparasitemic children with encephalopathy in a coma due to a cause other than malaria.
Circulating extracellular histones are elevated in CM. (A) Circulating histone levels measured in the serum of healthy controls (HC; n = 22), and patients with mild febrile illness (MF; n = 34), uncomplicated malaria (UM; n = 50), aparasitemic non-CM coma (Non-CM; n = 10) and admission serum from children who met the standard WHO case definition of CM at presentation (n = 218). Thick black lines and error bars are the geometric mean and 95% CI. (B) Refining the diagnosis of CM by retinal examination, comparison is made between circulating histone levels in children with Ret−CM (n = 48) and Ret+CM (n = 170). (C-F) Scatter plots to examine the correlation between histone levels and plasma factors in children with Ret+CM are shown. (C) Plasma fibrin monomer levels; (D) prothrombin fragment F1+2; (E) OPG; and (F) whole blood peripheral parasite density. (C-E) Data shown were measured by ELISA, and parasite density was calculated from white blood cell–normalized slide counts using thick smears. Comparison was made using ANOVA and Dunnett’s multiple comparison test (A), Student t test (B), and Pearson's correlation coefficient (C-F). Non-CM are aparasitemic children with encephalopathy in a coma due to a cause other than malaria.
To explore histones as a possible trigger for coagulation activation in CM, we assessed the association between circulating histones and markers of in vivo fibrin formation and coagulation activation.11 In Ret+CM cases, circulating histones correlated with plasma fibrin monomer concentrations (r = 0.56, P ≤ .001; Figure 1C) and weakly with prothrombin fragment F1+2 (a marker of thrombin generation; r = 0.34, P = .001; Figure 1D).
Endothelial activation with Weibel Palade body (WPB) exocytosis is considered a key component of malaria pathogenesis and promotes thrombosis.36,37 Soluble OPG is released from WPB into the blood and is a sensitive marker of WPB exocytosis correlating with development and severity of malaria.33,38 Here, circulating histone concentration correlated with plasma OPG concentration (r = 0.54, P < .001; Figure 1E).
The density of parasitemia alone does not account for these associations as there was minimal correlation between extracellular histone levels and peripheral parasite density (r = 0.20, P = .0044; Figure 1F) and there was no correlation between circulating histone levels and histidine-rich protein 2 levels (PfHRP2, a released parasite protein used as a marker of biomass [r = 0.09, P = .25]; data not shown). Further, fibrin monomer and OPG concentrations correlated less strongly with peripheral parasite density (r = 0.34, P ≤ .001 and r = 0.46, P ≤ .001) than with circulating histones. There was minimal correlation between fibrin monomers or OPG and PfHRP2 (r = 0.24, P = .013; r = 0.10, P = .34). Taken together, these data indicate an association between circulating histone levels and markers of endothelial activation, clot formation, and clot localization that is not explained by an association with parasite density.
Association between histone levels and brain swelling
Given the association between histones and endothelial processes implicated in vascular dysfunction in CM, we assessed the correlation between histone levels and fatal outcome and brain swelling. In Ret+CM cases, the geometric mean serum histone concentration on admission was not significantly different between those patients who later died (n = 24; geometric mean, 39.1 μg/mL; 95% CI, 19.2 to 79.6 μg/mL; Figure 2A) and patients who survived (n = 146; geometric mean, 22.8 μg/mL; 95% CI, 17.9 to 29.0 μg/mL; P = .11).
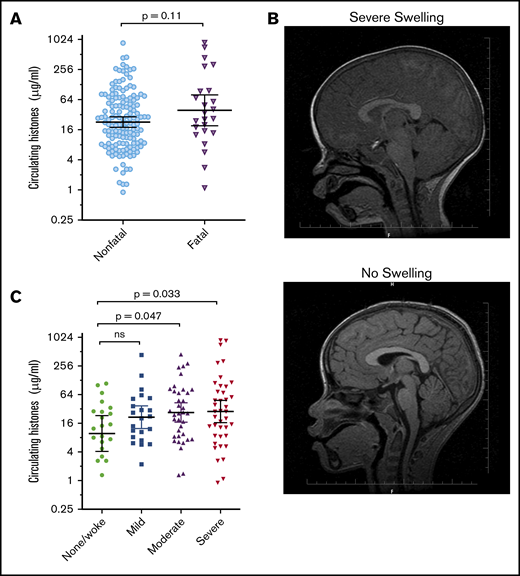
Extracellular histones are associated with the degree of brain swelling demonstrated on MRI scan. (A) Serum histone levels at admission in children who went on to die (Fatal; n = 24) and in those who survived (Nonfatal; n = 146). Differences between these groups were compared using a Student t test on log-transformed data. (B) Typical example of an MRI from a Malawian child with Ret+CM with a severely swollen brain (top) compared with an MRI from a Malawian child with Ret+CM with no swelling (bottom). (C) Circulating histones in children presenting with Ret+CM at different levels of brain swelling: those with no evidence of brain swelling or who woke up on the day of admission before they had a scan (n = 22) compared with mild (n = 22), moderate (n = 37), or severe brain swelling (n = 40). Black lines and error bars indicate geometric mean and 95% CI. Differences between the groups were compared by an ANOVA on log-transformed data with the Dunnett’s test to compare individual differences between no swelling and other swelling groups.
Extracellular histones are associated with the degree of brain swelling demonstrated on MRI scan. (A) Serum histone levels at admission in children who went on to die (Fatal; n = 24) and in those who survived (Nonfatal; n = 146). Differences between these groups were compared using a Student t test on log-transformed data. (B) Typical example of an MRI from a Malawian child with Ret+CM with a severely swollen brain (top) compared with an MRI from a Malawian child with Ret+CM with no swelling (bottom). (C) Circulating histones in children presenting with Ret+CM at different levels of brain swelling: those with no evidence of brain swelling or who woke up on the day of admission before they had a scan (n = 22) compared with mild (n = 22), moderate (n = 37), or severe brain swelling (n = 40). Black lines and error bars indicate geometric mean and 95% CI. Differences between the groups were compared by an ANOVA on log-transformed data with the Dunnett’s test to compare individual differences between no swelling and other swelling groups.
However, histone levels were nearly 3 times higher in children who had moderate brain swelling (geometric mean, 27.0 μg/mL; 95% CI, 16.9 to 43.2, P = .047) or severe brain swelling (geometric mean, 28.3 μg/mL; 95% CI, 16.5 to 48.5, P = .033; typical example Figure 2B, upper image) than in children who had no evidence of brain swelling on MRI (geometric mean, 9.8 μg/mL; 95% CI, 4.1 to 23.2; Figure 2B,C). Further, peripheral parasite density, PfHRP2, lactate, platelet count, and OPG levels were not significantly associated with brain swelling, at least among those patients for whom we had these data (supplemental Figure 4).
Approximately 50% of circulating histones are plasmodial
Owing to the highly conserved nature of histones, with >90% sequence homology between Plasmodium and human histones, available antibodies react with both human and Plasmodium histones.28 We developed a semiquantitative mass spectrometry method to determine the proportion of parasitic and human histones within patient samples (Figure 3A). Using P falciparum histones purified from culture and pure human histones, we identified specific peptides for both H4 (Figure 3B,C) and H2A that distinguished between P falciparum and human histones (Figure 3D,E). We then applied this method to serum samples from 10 children with Ret+CM to determine the ratio of P falciparum and human histones. P falciparum histones constituted a median of 50% of the total histones (interquartile range, 24.6% to 78.3%; Figure 3F,G). These are the first data demonstrating plasmodial histones in blood in patients.

Mass spectrometry analysis of origin of extracellular histones in CM cases. (A) Schematic representation of the methodology used for isolation, purification, and mass spectrometry analysis. Using Skyline software and by aligning trypsin fragments to reference amino acid sequences, we were able to identify specific histone H2A and H4 peptides that were present in purified P falciparum (malarial) preparations, that were not present in purified human histones (H1, H2A, H2B, H3, and H4) and vice versa. Typical peptides are presented from malarial (B) and human (C) database searches. Using Skyline software, we were able to identify histone H4 peptides for each species that demonstrated different mass/charge ratios with distinct human and P falciparum peptides and also distinct H2A human and P falciparum peptides (data not shown). This enabled us to identify, with high specificity, P falciparum (D) and human (E) species-specific peptides derived from histones spiked into PBS (left side of histogram, Histones) or serum (right side of histogram, Serum + Histones) Control is serum alone. Data shown are for H4. (F) In Ret+CM patient serum (n = 10) we were able to identify P falciparum histones H2A and H4 in the samples as well as human H2A and H4 (data not shown). (G) We combined the contribution of these 2 components to estimate the variable proportions of circulating human and P falciparum in the patient serum, demonstrating a significant contribution of P falciparum histones to the total pool.
Mass spectrometry analysis of origin of extracellular histones in CM cases. (A) Schematic representation of the methodology used for isolation, purification, and mass spectrometry analysis. Using Skyline software and by aligning trypsin fragments to reference amino acid sequences, we were able to identify specific histone H2A and H4 peptides that were present in purified P falciparum (malarial) preparations, that were not present in purified human histones (H1, H2A, H2B, H3, and H4) and vice versa. Typical peptides are presented from malarial (B) and human (C) database searches. Using Skyline software, we were able to identify histone H4 peptides for each species that demonstrated different mass/charge ratios with distinct human and P falciparum peptides and also distinct H2A human and P falciparum peptides (data not shown). This enabled us to identify, with high specificity, P falciparum (D) and human (E) species-specific peptides derived from histones spiked into PBS (left side of histogram, Histones) or serum (right side of histogram, Serum + Histones) Control is serum alone. Data shown are for H4. (F) In Ret+CM patient serum (n = 10) we were able to identify P falciparum histones H2A and H4 in the samples as well as human H2A and H4 (data not shown). (G) We combined the contribution of these 2 components to estimate the variable proportions of circulating human and P falciparum in the patient serum, demonstrating a significant contribution of P falciparum histones to the total pool.
Colocalization of parasite sequestration and luminal histone staining in cerebral microvessels in tissue from fatal CM cases
In mouse models of histone-induced vascular damage, histones bind to the luminal surface of the endothelium.20 To assess for the presence of extracellular histones at sites of sequestration and whether histones accumulate on the endothelial surface in CM, we performed immunohistochemical staining for histones in postmortem brain samples from Malawian children (details of individual cases in supplemental Table 1). Luminal histone staining was more frequent and strong in definitive CM cases (n = 17; Figure 4A, magnified in Figure 4B; supplemental Figure 7) than parasitemic non-CM cases who met the standard CM case definition while alive, but, at autopsy, had no sequestration in the brain and were found to have an alternative cause of coma and death (n = 6; Figure 4C; supplemental Figure 8) or nonmalarial comatose illness cases (n = 5; supplemental Figure 8).

Histones accumulate at the endothelial surface in the cerebral microvasculature and are associated with sequestration. (A,B) Fatal definitive CM cases showing histone staining in close proximity with endothelial cell luminal surface and marks host cell nuclei (red arrow) as well parasite nuclei in IE (white arrow). (B) Enlarged image of panel A to show luminal histone staining (black arrows) as well as IE with histone staining. (C) Fatal parasitemic non-CM comatose case (Parasitemic) with no histone endothelial membrane binding; histone staining can be seen in mammalian cell nuclei (red arrows). (D) Luminal histone staining is markedly increased in definitive CM (n = 17) compared with parasitemic non-CM cases (Parasitemic, n = 6) or fatal nonmalarial comatose illness CM cases (nonmalaria; n = 5). (E) In definitive CM cases (n = 17); there is a strong association between the degree of sequestration and the presence and strength of histone membrane staining (determined by ordinal logistic regression). Red staining is to antihistone H3 using Vector Red. Images were acquired at ×600 using an oil immersion lens.
Histones accumulate at the endothelial surface in the cerebral microvasculature and are associated with sequestration. (A,B) Fatal definitive CM cases showing histone staining in close proximity with endothelial cell luminal surface and marks host cell nuclei (red arrow) as well parasite nuclei in IE (white arrow). (B) Enlarged image of panel A to show luminal histone staining (black arrows) as well as IE with histone staining. (C) Fatal parasitemic non-CM comatose case (Parasitemic) with no histone endothelial membrane binding; histone staining can be seen in mammalian cell nuclei (red arrows). (D) Luminal histone staining is markedly increased in definitive CM (n = 17) compared with parasitemic non-CM cases (Parasitemic, n = 6) or fatal nonmalarial comatose illness CM cases (nonmalaria; n = 5). (E) In definitive CM cases (n = 17); there is a strong association between the degree of sequestration and the presence and strength of histone membrane staining (determined by ordinal logistic regression). Red staining is to antihistone H3 using Vector Red. Images were acquired at ×600 using an oil immersion lens.
Quantifying this by scoring (scorer comparison shown in supplemental Figure 9), histone staining was stronger in definitive CM cases compared with the non-CM parasitemic cases (parasitemic, described as CM3 in previous studies1 ; Figure 4D; odds ratio [OR], 2.6; 95% CI, 1.7 to 3.9; P < .001) or nonmalarial cases (nonmalaria; OR, 7.2; 95% CI, 5.0 to 10.6; P < .001). Among definitive CM cases, there was a strong association between histone luminal staining and the presence of parasite sequestration (Figure 4E). When sequestration was present but in less than 50% of the vessel (+) the OR of histone membrane staining being present was 5.2 (95% CI, 2.8 to 9.7; P < .001); when greater than 50% of the vessel contained sequestered parasites (++) the OR for the presence of histone staining was 16.9 (95% CI, 9.2 to 31.3; P < .001; Figure 4E). The accumulation of luminal histones in association with parasite sequestration supports a parasite origin for bound histones, although this could also indicate that sequestration induces local production host histones (eg, through neutrophil extracellular trap [NET] formation39,40 or cellular damage).
High histone levels are associated with BBB breakdown and thrombosis in tissue from fatal CM cases
Histone staining was also associated with areas of BBB breakdown, demonstrated by staining for fibrinogen extravasation (Figure 5A). Weak histone staining was associated with an OR of 2.8 for the presence of fibrinogen extravasation (95% CI, 1.6 to 5.0; P ≤ .001) and strong histone staining with an OR of 4.5 for fibrinogen extravasation (95% CI, 1.8 to 11.4; P = .001), as shown in Figure 5B as the percentage of vessels with a leak. This was not simply explained by histone staining being associated with sequestration, as the correlation between histone staining and leak remained significant when the degree of sequestration was included as a covariate in the regression model (revised OR, 2.6; 95% CI, 1.4 to 4.8; P = .002 for weak histone staining and OR, 5.1; 95% CI, 1.9 to 13.5; P = .001 for strong staining). Histone staining was also observed to colocalize with thrombi (Figure 5C) and with ring hemorrhages (Figure 5D). These data implicate histones as potential mediators of endothelial disruption at sites of sequestration in the brain, leading to BBB breakdown and thrombosis.
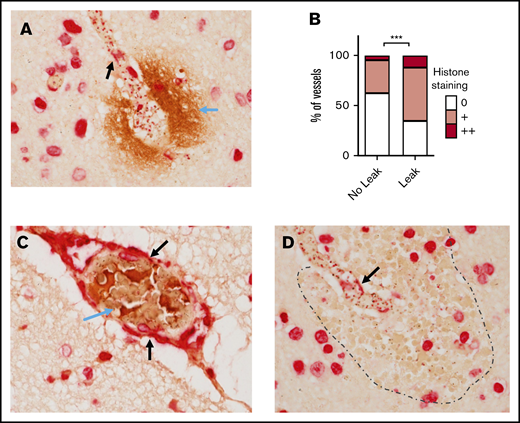
Luminal bound histones colocalize with areas of BBB breakdown and coagulopathy. (A) Histone endothelial membrane staining (black arrows) colocalizing with fibrinogen extravasation (blue arrow), which is indicative of BBB breakdown. (B) Strong association between the extent of histone endothelial membrane staining and the presence of fibrinogen extravasation, determined by ordinal logistic regression. (C) Histone membrane staining (black arrows) colocalizing with thrombosis (blue arrow). (D) Histone membrane staining (black arrow) colocalizing with a ring hemorrhage (edge demarcated by dotted line). Red staining is to anti-H3 antibody using Vector Red and brown staining is to antifibrinogen antibody using 3,3′-diaminobenzidine. Images were acquired at ×600 using an oil immersion lens.
Luminal bound histones colocalize with areas of BBB breakdown and coagulopathy. (A) Histone endothelial membrane staining (black arrows) colocalizing with fibrinogen extravasation (blue arrow), which is indicative of BBB breakdown. (B) Strong association between the extent of histone endothelial membrane staining and the presence of fibrinogen extravasation, determined by ordinal logistic regression. (C) Histone membrane staining (black arrows) colocalizing with thrombosis (blue arrow). (D) Histone membrane staining (black arrow) colocalizing with a ring hemorrhage (edge demarcated by dotted line). Red staining is to anti-H3 antibody using Vector Red and brown staining is to antifibrinogen antibody using 3,3′-diaminobenzidine. Images were acquired at ×600 using an oil immersion lens.
Histones purified from P falciparum and sera from Ret+CM patients cause barrier disruption and are toxic to cultured brain EC
Mammalian histones cause barrier disruption and are toxic to human umbilical vein EC.16,20 P falciparum histones induce barrier disruption and are toxic to dermal and lung EC.28 We studied the effects of exposing primary HBMEC to purified plasmodial histones, resulting in rapid barrier disruption that was reversed by antihistone single-chain fragment variable (ahscFv; Figure 6A).20,26 Serum from patients with Ret+CM induced significant (more than threefold) increase in barrier disruption, whereas serum from Ret−CM, UM, and non-CM coma patients did not (Figure 6B). This barrier disruption was abrogated by specific blocking of histone activity in serum with ahscFv (Figure 6C). In a real-time impedance system, TEER was reduced by application of purified plasmodial histones (Figure 6D,E; supplemental Figure 10A). This reduction was abrogated by nonanticoagulant heparin (a potential treatment with minimal toxicity that prevents toxicity of mammalian histones27 ; Figure 6F; supplemental Figure 10A) or by specific removal of histones using ahscFv-coated beads (supplemental Figure 10B,C). Purified histones induced toxicity on HBMEC in a dose-dependent manner (supplemental Figure 11A) abrogated by ahscFv (supplemental Figure 11B) and heparin (supplemental Figure 11C). Serum from Ret+CM cases (n = 13) were more toxic than histones from patients with Ret−CM (n = 5), non-CM coma (n = 3), uncomplicated malaria (n = 5), or nonmalarial febrile illness (n = 5; Figure 6G). Considering that this toxicity might be histone related, when we segregated Ret+CM cases by histone levels, those with elevated histones (>100 μg/mL, n = 8) induced significantly more cellular toxicity than Ret+CM cases with lower histone levels (<25 g/mL, n = 5; Figure 6H). This toxicity was reversed by ahscFv (Figure 6I) and by nonanticoagulant heparin (Figure 6J). Taken together, these data indicate that histones cause barrier disruption and toxicity to brain EC, and histones present in Ret+CM serum are active and necessary for causing serum-induced barrier disruption and toxicity to brain EC ex vivo. Considering this as a possible therapeutic target, this toxicity can be prevented by nonanticoagulant heparin.
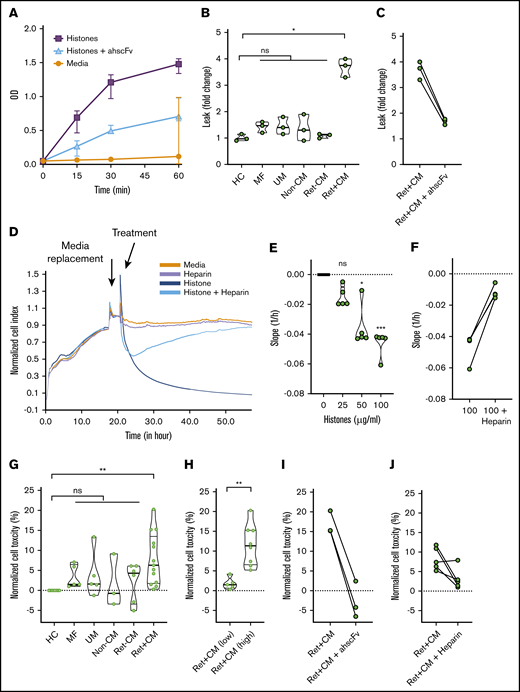
P falciparum histones are toxic factors in CM patient sera which are necessary to induce brain endothelial disruption ex vivo. (A) Time course of barrier disruption of primary HBMEC by P falciparum histones in a Transwell dual chamber system treated with 100 μg/ml of purified plasmodial histones compared with cells with media alone or cells treated with histones plus 200 μg/ml of antihistone single chain variable Fragment (ahscFv). Barrier disruption was assessed by measuring the optical density (OD) of the liquid in the lower part of the Transwell system (measures horseradish peroxidase pass through, detected by reaction with tetramethylbenzidine substrate, 3 biological replicates per condition). By 2-way ANOVA there was significant barrier disruption in the histone group when compared with the media group (P = .0045). This was abrogated by treatment with ahscFv, which was not significantly different from the media-only group (P = .093). (B) Serum-induced barrier disruption by patient samples. Transwell permeability changes of HBMEC monolayer are expressed as fold changes in horseradish peroxidase pass-through over 1 hour. Data are expressed as violin plots and analyzed with the Kruskal-Wallis test with Dunn’s test to adjust for multiple comparisons. Compared with healthy control serum there was a significant increase in permeability induced by the patient serum from Ret+CM cases (P = .0123), but not with any of the other patient groups (n = 3 biological replicates for all conditions): mild febrile illness (MF; P = .423), uncomplicated malaria (UM; P = .580), non-CM comatose illness (non-CM; P > .99), and Ret−CM (P > .99). (C) Histone-induced disruption is abrogated by ahscFv (200 μg/ml, P = .44 when compared with media-only condition). (D) HBEC were grown on gold-electrode–coated plates in an impedance system (xCELLigence) that enables real-time noninvasive measurement of TEER. Confluent cells were treated with purified P falciparum histones with or without heparin (200 μg/ml) or histones preincubated with magnetic beads coated with ahscFv (200 μg/ml), with beads removed by magnet prior to incubation. (E) The effect of histone concentration on barrier disruption was measured as downward slope, reflecting changes in cell impedance. Compared with the media-only control there was no significant difference at 25 µg/ml histone (P = .35); however, significant disruption was observed at histone concentrations of 50 µg/ml (P = .011) and 100 µg/ml (P = .0009) (Kruskal-Wallis test with Dunn’s test). (F) These differences were abrogated by prior treatment with heparin (P = .23). (G) HBMECs were treated for 1 hour with serum from patients with or without ahscFv or nonanticoagulant heparin (200 μg/ml). Cell toxicity was determined by propidium iodide staining using flow cytometry. Data are expressed as medians and IQR relative to cells treated with media alone (set to 0%) and compared using the Kruskal-Wallis with Dunn’s test to adjust for multiple comparisons. Compared with serum from the healthy control (HC; n = 8), serum from Ret+CM cases (n = 13) induced significant toxicity (P = .0012), whereas serum from other patient groups did not: UM (n = 5; P = .50), MF (n = 5; P = .23), non-CM (n = 5; P = .99), Ret−CM; (n = 6; P = .69). (H) Among Ret+CM cases, serum from cases with histones >100 μg/ml (n = 8) caused higher toxicity than those with histone <25 μg/ml (n = 5, P = .0016,); Mann-Whitney U test. (D) This was abrogated by treatment with ahscFv (n = 3, P = .205) or heparin (n = 5, P = .115).
P falciparum histones are toxic factors in CM patient sera which are necessary to induce brain endothelial disruption ex vivo. (A) Time course of barrier disruption of primary HBMEC by P falciparum histones in a Transwell dual chamber system treated with 100 μg/ml of purified plasmodial histones compared with cells with media alone or cells treated with histones plus 200 μg/ml of antihistone single chain variable Fragment (ahscFv). Barrier disruption was assessed by measuring the optical density (OD) of the liquid in the lower part of the Transwell system (measures horseradish peroxidase pass through, detected by reaction with tetramethylbenzidine substrate, 3 biological replicates per condition). By 2-way ANOVA there was significant barrier disruption in the histone group when compared with the media group (P = .0045). This was abrogated by treatment with ahscFv, which was not significantly different from the media-only group (P = .093). (B) Serum-induced barrier disruption by patient samples. Transwell permeability changes of HBMEC monolayer are expressed as fold changes in horseradish peroxidase pass-through over 1 hour. Data are expressed as violin plots and analyzed with the Kruskal-Wallis test with Dunn’s test to adjust for multiple comparisons. Compared with healthy control serum there was a significant increase in permeability induced by the patient serum from Ret+CM cases (P = .0123), but not with any of the other patient groups (n = 3 biological replicates for all conditions): mild febrile illness (MF; P = .423), uncomplicated malaria (UM; P = .580), non-CM comatose illness (non-CM; P > .99), and Ret−CM (P > .99). (C) Histone-induced disruption is abrogated by ahscFv (200 μg/ml, P = .44 when compared with media-only condition). (D) HBEC were grown on gold-electrode–coated plates in an impedance system (xCELLigence) that enables real-time noninvasive measurement of TEER. Confluent cells were treated with purified P falciparum histones with or without heparin (200 μg/ml) or histones preincubated with magnetic beads coated with ahscFv (200 μg/ml), with beads removed by magnet prior to incubation. (E) The effect of histone concentration on barrier disruption was measured as downward slope, reflecting changes in cell impedance. Compared with the media-only control there was no significant difference at 25 µg/ml histone (P = .35); however, significant disruption was observed at histone concentrations of 50 µg/ml (P = .011) and 100 µg/ml (P = .0009) (Kruskal-Wallis test with Dunn’s test). (F) These differences were abrogated by prior treatment with heparin (P = .23). (G) HBMECs were treated for 1 hour with serum from patients with or without ahscFv or nonanticoagulant heparin (200 μg/ml). Cell toxicity was determined by propidium iodide staining using flow cytometry. Data are expressed as medians and IQR relative to cells treated with media alone (set to 0%) and compared using the Kruskal-Wallis with Dunn’s test to adjust for multiple comparisons. Compared with serum from the healthy control (HC; n = 8), serum from Ret+CM cases (n = 13) induced significant toxicity (P = .0012), whereas serum from other patient groups did not: UM (n = 5; P = .50), MF (n = 5; P = .23), non-CM (n = 5; P = .99), Ret−CM; (n = 6; P = .69). (H) Among Ret+CM cases, serum from cases with histones >100 μg/ml (n = 8) caused higher toxicity than those with histone <25 μg/ml (n = 5, P = .0016,); Mann-Whitney U test. (D) This was abrogated by treatment with ahscFv (n = 3, P = .205) or heparin (n = 5, P = .115).
Discussion
Histones are nuclear proteins that can be released from damaged host cells and have been shown to be released during the rupture of schizonts.28 Here we demonstrate that circulating histones are elevated in patients with CM. Specifically, P falciparum histones are present in the blood of children with Ret+CM, with circulating histone levels associated with endothelial and coagulation activation and with brain swelling. Luminal histone staining in postmortem brain tissue colocalizing with sequestration and their independent association with leak indicate a potential role for histones in sequestration-driven BBB breakdown. We highlight potential translational relevance; the toxicity of serum from Ret+CM cases can be inhibited by antihistone reagents including nonanticoagulant heparin.
A number of factors released from IE have been shown to cause endothelial damage or leak in vitro,41 including glycosylphosphatidylinositol,42 extracellular vesicles,43 heme,40,44 and PfHRP2.45 IE–EC receptor–ligand interactions also cause endothelial perturbation.46-48 Although it seems likely that CM pathogenesis consists of a combination of interacting factors rather than a single toxin or ligand,49,50 we sought a factor that is necessary for CM vascular pathology and has therapeutic potential. Hallmark features of extracellular histones in other critical illnesses are microvascular leak and thrombosis at sites of histone release, which are both considered key components of CM pathogenesis and are notably concentrated at sites of IE sequestration. This is in keeping with the anticipated concentration of histone release at sites of sequestration in the brain from histone-packed schizonts. Histones are a plausible target for an adjunctive therapy, because antihistone reagents are protective in animal models of sepsis,16 acute lung injury,20 and pancreatitis.51
Consistent with the release of P falciparum histones in patients with malaria, nucleosomes have been detected in the plasma of South-East–Asian adults with malaria, and were higher in severe cases.28 However the association between nucleosomes (which have minimal toxicity24 ) and free histones is variable, and it was not identified whether these nucleosomes were of host or parasite origin or whether they were active. In this study, we have demonstrated that high levels of circulating histones released from both parasites and host cells in CM correlate with the level of brain swelling on MRI, suggesting that both parasite and host histones in the circulation may play a role in BBB breakdown and leakage. Treating human brain EC with serum from CM patients or isolated parasite histones further supported their roles in brain endothelial damage and barrier disruption.
The colocalization of parasite sequestration and of luminal histone staining with thrombosis and fibrinogen extravasation in postmortem brain tissue support that parasite histones may trigger these processes. The rupture of sequestered schizonts would be predicted to result in elevated levels of extracellular histones locally. Histones promote coagulation activation,52 thrombosis,36 and endothelial disruption16,20 which could, in turn, culminate in BBB breakdown (hypothesized mechanism summarized in supplemental Figure 12).
The typical observed clinical pattern of disease in CM in African children is of multifocal microvascular leakage and thrombosis in the neurovasculature with resultant brain swelling, associated clinically with deep coma but without multiorgan failure14 or overt disseminated intravascular coagulation.11 A key consideration is how extracellular histones, which in some cases reach levels in the circulation that are capable of inducing cell toxicity and barrier disruption, induce leak preferentially in the brain. It is notable that the geometric mean concentration of histones in the serum in Ret+CM cases was 25 μg/mL, but significant reductions of HBMEC viability in our assay were only seen at histone concentrations greater than 50 μg/mL (similar to the toxicity threshold for mammalian histones16,20,28 ). The implication is that, in most patients with CM, histone levels in the circulation do not reach levels sufficient to cause systemic toxicity. In contrast, intense sequestration and an increase in nuclear material in sequestered schizonts by up to 30-fold would be predicted to massively concentrate release of P falciparum histones at the endothelial surface. Thus, at sites of intense sequestration (Figure 4A,B), histone exposure may overwhelm homeostatic mechanisms and cross this toxic threshold, leading to focal endothelial break down and localized intravascular coagulation (supplemental Figure 12). The balance between homeostasis and decompensation is likely to vary between individuals and between different vascular beds depending on the degree of histone production and on local capacity to break them down.
The brain may be particularly vulnerable to these mechanisms. First, there are high levels of sequestration in the brain in CM.14,53,54 Second, the human brain may have reduced capacity to breakdown histones because it has reduced innate capacity to produce aPC,55 owing to low constitutive thrombomodulin and endothelial protein C receptor (EPCR) expression,56,57 the receptors involved in aPC production. Moreover, IE sequestration itself causes EPCR shedding, and parasite variants associated with the development of CM use EPCR as a binding receptor,58,59 interfering with its function and the production of aPC.59,60 Thus, histones released by IE would be predicted to concentrate and be particularly detrimental in the brain.
Our study has several limitations. First, it is in human patients. Although generally a strength, this leads to marked heterogeneity, including in variables that might affect histone levels, such as length of illness and timing of antimalarial drug administration. Further, we collected blood samples from each patient at only 1 time point, representing a snapshot in a dynamic disease process. This precluded examination of the temporal association between histone levels and other variables. Second, although the association between histone binding and sequestration and the finding that ∼50% of histones in serum were of parasite origin are both suggestive of a parasite origin for luminal histones, we did not prove this. Nonetheless, concentration of host histones at sites of IE sequestration (eg, through NETs39,40 ) would also be predicted to have similar effects and to respond to similar treatments.
Further work is warranted to explore the biology and timing of plasmodial histone release and to explore the therapeutic potential of antihistone reagents. Modified nonanticoagulant heparins are a rational first choice given their use in critically ill patients with a variety of inflammatory diseases61 and sickle cell crisis.62 Heparins were historically used for treatment of severe and CM63 with some evidence of efficacy,64 but their use was discontinued because of concerns of inducing bleeding.65 There has been renewed interest in the use of heparin following development of analogs with reduced anticoagulant activity.61,66 These modified heparins can disrupt IE sequestration67,68 and rosetting67,69-71 and block merozoite invasion.66,72,73 A modified nonanticoagulant heparin (Sevuparin) was shown to safely reduce parasite invasion and release mature stage parasites.74 Larger studies are planned to assess efficacy at reducing parasite biomass and sequestration. Further investigation is required to determine whether a different dosing regimen is needed to reverse the effects of histones rather than to decrease merozoite invasion and sequestration. However, the possibility that modified heparins could be synergistic in malaria both by reducing the density of circulating and sequestered parasites and by neutralizing the effect of histones, make the potential benefits more compelling.
Send data sharing requests via e-mail to the corresponding author, Christopher A. Moxon (christopher.moxon@glasgow.ac.uk).
Acknowledgments
The authors thank the nurses and clinicians on the Pediatric Research Ward team (Malawi-Liverpool-Wellcome Clinical Research Program and Blantyre Malaria Project) and the nurses and clinicians in the Department of Pediatrics and Child Health (Queen Elizabeth Hospital, Blantyre, Malawi) for recruiting and caring for patients. The authors thank C. Powell (Roald Dahl Haemostasis & Thrombosis Centre, Liverpool, United Kingdom) for performing the fibrin-based assays. The authors also thank Dan Milner (American Society for Clinical Pathology) for histopathology advice; Qian Zhen (Program in Dermatopathology, Department of Pathology, Brigham and Women's Hospital, Boston, MA) for technical support with histopathology; Dyann Wirth for providing laboratory space and advice; and Andy Waters and Matthias Marti (Wellcome Centre for Integrative Parasitology, University of Glasgow) for helpful comments on the manuscript.
This work was supported by funding from the Wellcome Trust (109698/Z/15/Z) (C.A.M.) and Academy of Medical Sciences (C.A.M.), and grants from the National Institutes of Health, National Institute of Allergy and Infectious Diseases (5R01AI034969-14) (T.E.T.) and the British Heart Foundation (PG/14/19/30751 and PG/16/65/32313) (G.W.). The Malawi–Liverpool Wellcome Clinical Research Programme is supported by core funding from the Wellcome Trust (084679/Z/08/Z). The Wellcome Centre for Integrative Parasitology is supported by core funding from the Wellcome Trust (104111).
Authorship
Contribution: C.A.M., S.T.A., A.G.C., G.W., and C.-H.T. conceived the study and designed the experiments; C.A.M., S.L., D.M., and S.T.A. performed analysis; Y.A., S.T.A., J.Y.K., J.S., J.M.H.T., N.O., D.M., and C.A.M. performed laboratory experiments; K.B.S. and T.E.T. ran the clinical study and provided clinical and scientific input; M.M. provided clinical and scientific input; G.M., G.G.-C., and J.S.O. designed experiments and provided scientific input; S.K. and M.P. analyzed the MRI scans; C.A.M. wrote the original draft with significant input from A.G.C, S.T.A., and C.-H.T.; and all authors contributed to the critical review and editing of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Christopher A. Moxon, Wellcome Centre for Integrative Parasitology, Institute of Infection, Immunity and Inflammation, College of Medical, Veterinary and Life Sciences, University of Glasgow, Sir Graeme Davies Building, 120 University Place, Glasgow G12 8TA, United Kingdom; e-mail: christopher.moxon@glasgow.ac.uk.

