

Key Points
TCR clones can be followed throughout the disease course with deep NGS sequencing of the TCR Vβ CDR3 and STAT3.

Abstract
T large granular lymphocyte leukemia (T-LGLL) is a clonal lymphoproliferative disorder that can arise in the context of pathologic or physiologic cytotoxic T-cell (CTL) responses. STAT3 mutations are often absent in typical T-LGLL, suggesting that in a significant fraction of patients, antigen-driven expansion alone can maintain LGL clone persistence. We set out to determine the relationship between activating STAT3 hits and CTL clonal selection at presentation and in response to therapy. Thus, a group of patients with T-LGLL were serially subjected to deep next-generation sequencing (NGS) of the T-cell receptor (TCR) Vβ complementarity-determining region 3 (CDR3) and STAT3 to recapitulate clonal hierarchy and dynamics. The results of this complex analysis demonstrate that STAT3 mutations produce either a sweeping or linear subclone within a monoclonal CTL population either early or during the course of disease. Therapy can extinguish a LGL clone, silence it, or adapt mechanisms to escape elimination. LGL clones can persist on elimination of STAT3 subclones, and alternate STAT3-negative CTL clones can replace therapy-sensitive CTL clones. LGL clones can evolve and are fueled by a nonextinguished antigenic drive. STAT3 mutations can accelerate this process or render CTL clones semiautonomous and not reliant on physiologic stimulation.
Introduction
Characterized by an increased number of circulating clonal cytotoxic T cells (CTLs), large granular T lymphocyte leukemia (T-LGLL) is frequently accompanied by neutropenia, anemia, or thrombocytopenia, but it often develops silently, without pathologic features or clinical symptoms.1-5 Many lines of evidence indicate that LGL clonal expansion evolves in the context of physiologic (viruses, tumor surveillance) or pathologic responses (autoimmune conditions) to antigens that drive excessive clonal expansions.6 This view has been supported by studies of T-cell receptor (TCR) rearrangement and TCR Vβ complementarity-determining region 3 (CDR3) deep sequencing, which has frequently demonstrated extreme expansion of a single immunodominant T-cell clone or multiple codominant clones.7-10 Discovery of activating STAT3 mutations in a significant proportion of patients with LGL suggested that such mutations initiated a clonal process yielding a spectrum of hematologic pathologies dependent on the specificity of the transformed/expanded CTL clones. Vβ CDR3 sequences acting as biological bar codes can be used to identify and quantify these expansions and to diagnose LGL.11-17
Deep TCR repertoire sequencing can precisely identify and assess CDR3 diversity and quantify immunodominant expansions. Using it in conjunction with next-generation sequencing (NGS) of STAT3 allows serial monitoring of clonal burden and reconstruction of the process that culminates in LGL. We used this approach to recapitulate the dynamics of clonal CTL expansions and interactions between individual clones in a large cohort of patients with T-LGLL followed over extended periods of time. Our goal was to determine whether poly- and subsequently oligoclonal immune responses are “anchored” by STAT3 mutations, most likely occurring in the most proliferative clones, or whether the responses were driven by somatic hits in STAT3 as the ancestral event. We also set out to characterize the plasticity of the underlying immune response during the course of treatment, including mechanisms of resistance or relapse.
Methods
DNA samples
Blood samples were obtained from patients with LGL seen at the Cleveland Clinic, according to protocols approved by the Cleveland Clinic institutional review board and the Declaration of Helsinki (for diagnostic criteria, see supplemental Table 1).15,18 DNA was extracted from blood mononuclear cells; the same sample was used for both sequencing methods. Samples were collected from a total of 207 patients with LGL seen at the Cleveland Clinic, 92% T-LGLL, and 8% natural killer LGL. Most patients presented with anemia (46%) and/or neutropenia (46%). Vβ expansion (by Vβ flow cytometry) was present in 94% of cases, with an average LGL count of 2317 k/μL. Clinical features are described in detail in supplemental Table 2.
Deep STAT3 NGS
All patients with LGL were deep sequenced for the presence of a mutation in exon 21 of STAT3, the protein-protein interaction domain.12 The average coverage was 7500 ± 5700 reads, and hits more than 1% were positive. A STAT3 mutation was found in 38% of patients in 4 common hotspots: 42% Y640F, 34% D661Y, 11% D661V, and 8% N647I. STAT3MT VAF was followed over multiple points for 44% of the total cohort. Multiple STAT3MT were found in 4% of patients. Whole-exome sequencing was performed for some patients, but no recurrent somatic mutations beyond STAT3 were found.
Deep TCR sequencing
TCR Vβ was sequenced using the Immunoseq assay (Adaptive Biotechnologies), which targets the TCRβ CDR3 region using a multiplex polymerase chain reaction library preparation.8 Libraries were sequenced on the MiSeq (Illumina), with an average of 40 773 ± 35 667 productive templates. The nucleotide and predicted amino acid sequences, along with the rearranged VDJ regions, were available for every unique CDR3 sequence. TCR deep sequencing was performed on 23% of patients, 10% of which were sequenced at more than 1 point. The diversity of the same represents how monoclonal or polyclonal a sample is as previously defined; values approaching 0 reflect extreme monoclonal samples.8
Results
Compared with healthy control patients, the diversity of patients with LGL is much smaller, and they have a higher average max Vβ clone size, suggesting they are more monoclonal (Figure 1). Starting from an initial cohort of 207 patients with LGLL, we selected a representative 18 T-LGLL cases to deep sequence both STAT3 and TCRB CDR3 longitudinally (Table 1). Patients were sequenced for an average of 4 times (range, 2-8 times). In approximately half the patients, we were able to detect STAT3MT in previously described canonical positions.13-15 In all patients, deep TCR NGS identified at least 1 clonotype that was immunodominant over most other clonotypes, with multiple immunodominant subclonotypes identified in about half the patients. For control purposes, we also longitudinally sequenced 2 patients with natural killer LGL; major TCR clonotypic expansions could not be detected in 1 patient with a STAT3 mutation, but 2 immunodominant T-cell clones were identified in the other patient, consistent with sting antigenic drive also involving T-cell responses (supplemental Figure 1).
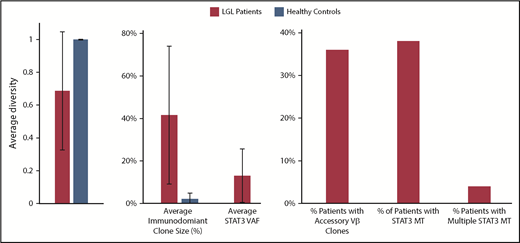
Characterization of LGL and healthy control patients by deep TCR Vβ sequencing. A diversity value of 1 represents a polyclonal sample.
Characterization of LGL and healthy control patients by deep TCR Vβ sequencing. A diversity value of 1 represents a polyclonal sample.
Longitudinal T-LGLL characteristics of patients with leukemia
| STAT3MT | Hematologic presentation | AAD, y | Sex | Therapy | LGL count* | Flow cytometry | TCR deep sequencing | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VB ID | Clone size*, % | Major clonotype(s) | Clone size*, % | Clonal diversity*,† | Clonality*,† | |||||||
| CCF1 | D661V | Anemia | 68 | M | CSA, CYC, MTX | 2455 | NIP | 65 | CASSLLAGGYNEQFF | 42 | 0.176 | 0.373 |
| CCF2 | D661V | Neutropenia thrombocytopenia | 81 | M | MTX | 1225 | TRBV9 | 72 | CASSVGQGSPLHF | 75 | 0.565 | 0.734 |
| CCF3 | Y640F | Neutropenia thrombocytopenia | 55 | F | CSA | 1355 | TRBV6-2, TRBV3-1 | 25, 16 | CASSLASISYNEQFF | 26 | 0.068 | 0.263 |
| CCF4 | D661Y, Y640F | Pancytopenia | 64 | M | CSA, MTX | 3672 | TRBV10-3 | 23 | CASSAPREGTAYEQYF, CASSPTSGGEQFF | 8, 5 | 0.016 | 0.222 |
| CCF5 | D661Y | Anemia, neutropenia | 54 | M | CSA, CYC | 10 631 | TRBV9 | 99 | CASSVGRFQETQYF | 44 | 0.192 | 0.378 |
| CCF6 | Y640F | Neutropenia | 47 | M | CSA, CYC, MTX | 850 | TRBV29-1 | 8, 11 | CASSTFAEIRQPQHF, CASSLTENYGYTF | 4, 0.01 | 0.002 | 0.046 |
| CCF7 | D661Y | Neutropenia | 40 | M | CSA, CYC, MTX | 2882 | NIP | 95 | CATSRGTGDSYEQYF | 97 | 0.934 | 0.938 |
| CCF8 | Y640F | Anemia, neutropenia | 37 | F | CSA | 480 | TRBV12-3, TRBV12-4 | 82 | CASSSLTGLPPYEQYF | 29 | 0.085 | 0.247 |
| CCF9 | Y640F | Anemia, neutropenia | 74 | F | CSA, CYC, MTX | 496 | NIP | 96 | CASSPEGFQPQHF, CASTSFGTGTDTQYF | 91, 0.16 | 0.819 | 0.871 |
| CCF10 | Negative | Anemia, neutropenia | 17 | M | CSA, MTX | 330 | TRBV18 | 77 | CASSPFWTLEKLFF | 93 | 0.871 | 0.893 |
| CCF11 | Negative | Anemia | 63 | M | CSA, CYC, MTX | 980 | TRVB28, TRBV19 | 32, 18 | CASSPLGAVGYNEQFF, CAISEFPGGQTSDEGFF | 25, 1 | 0.086 | 0.471 |
| CCF12 | Negative | Neutropenia | 47 | F | CSA, MTX | 188 | TRBV12-3, TRBV12-4 | 45 | CASSIAGLGSEQFF, CASSEGQMYNEQFF | 12, 3 | 0.019 | 0.191 |
| CCF13 | Negative | Anemia, neutropenia | 52 | F | CSA, CYC, MTX | 261 | TRBV13 | 97 | CASSDLAGMLGRQFF | 99 | 0.983 | 0.984 |
| CCF14 | Negative | Anemia | 66 | M | CYC | 409 | NIP, TRBV20-1 | 84, 24 | CASSLEIAGGLIYNEQFF, CASSPGLAGGRETQYF | 27, 2 | 0.093 | 0.350 |
| CCF15 | Negative | Anemia, neutropenia | 60 | M | CSA, CYC, MDX | 4472 | TRBV19 | 97 | CASSIGIQPQHF | 83 | 0.686 | 0.791 |
| CCF16 | Negative | Asymptomatic | 59 | M | SCT, CYC | 1515 | TRBV27 | 30 | CASSSVGYEQYF | 40 | 0.173 | 0.478 |
| CCF17 | Negative | Anemia | 71 | F | MTX | 2536 | TRBV19 | 20 | CASRASGSDEQYF, CARSRDKGTDYGYTF | 10, 8 | 0.024 | 0.263 |
| CCF18 | Negative | Asymptomatic | 54 | M | None | 686 | TRBV27, TRVB4-3 | 12, 8 | CASSLGNYEQYF, CASSISQNTEAFF | 4, 7 | 0.026 | 0.319 |
| STAT3MT | Hematologic presentation | AAD, y | Sex | Therapy | LGL count* | Flow cytometry | TCR deep sequencing | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VB ID | Clone size*, % | Major clonotype(s) | Clone size*, % | Clonal diversity*,† | Clonality*,† | |||||||
| CCF1 | D661V | Anemia | 68 | M | CSA, CYC, MTX | 2455 | NIP | 65 | CASSLLAGGYNEQFF | 42 | 0.176 | 0.373 |
| CCF2 | D661V | Neutropenia thrombocytopenia | 81 | M | MTX | 1225 | TRBV9 | 72 | CASSVGQGSPLHF | 75 | 0.565 | 0.734 |
| CCF3 | Y640F | Neutropenia thrombocytopenia | 55 | F | CSA | 1355 | TRBV6-2, TRBV3-1 | 25, 16 | CASSLASISYNEQFF | 26 | 0.068 | 0.263 |
| CCF4 | D661Y, Y640F | Pancytopenia | 64 | M | CSA, MTX | 3672 | TRBV10-3 | 23 | CASSAPREGTAYEQYF, CASSPTSGGEQFF | 8, 5 | 0.016 | 0.222 |
| CCF5 | D661Y | Anemia, neutropenia | 54 | M | CSA, CYC | 10 631 | TRBV9 | 99 | CASSVGRFQETQYF | 44 | 0.192 | 0.378 |
| CCF6 | Y640F | Neutropenia | 47 | M | CSA, CYC, MTX | 850 | TRBV29-1 | 8, 11 | CASSTFAEIRQPQHF, CASSLTENYGYTF | 4, 0.01 | 0.002 | 0.046 |
| CCF7 | D661Y | Neutropenia | 40 | M | CSA, CYC, MTX | 2882 | NIP | 95 | CATSRGTGDSYEQYF | 97 | 0.934 | 0.938 |
| CCF8 | Y640F | Anemia, neutropenia | 37 | F | CSA | 480 | TRBV12-3, TRBV12-4 | 82 | CASSSLTGLPPYEQYF | 29 | 0.085 | 0.247 |
| CCF9 | Y640F | Anemia, neutropenia | 74 | F | CSA, CYC, MTX | 496 | NIP | 96 | CASSPEGFQPQHF, CASTSFGTGTDTQYF | 91, 0.16 | 0.819 | 0.871 |
| CCF10 | Negative | Anemia, neutropenia | 17 | M | CSA, MTX | 330 | TRBV18 | 77 | CASSPFWTLEKLFF | 93 | 0.871 | 0.893 |
| CCF11 | Negative | Anemia | 63 | M | CSA, CYC, MTX | 980 | TRVB28, TRBV19 | 32, 18 | CASSPLGAVGYNEQFF, CAISEFPGGQTSDEGFF | 25, 1 | 0.086 | 0.471 |
| CCF12 | Negative | Neutropenia | 47 | F | CSA, MTX | 188 | TRBV12-3, TRBV12-4 | 45 | CASSIAGLGSEQFF, CASSEGQMYNEQFF | 12, 3 | 0.019 | 0.191 |
| CCF13 | Negative | Anemia, neutropenia | 52 | F | CSA, CYC, MTX | 261 | TRBV13 | 97 | CASSDLAGMLGRQFF | 99 | 0.983 | 0.984 |
| CCF14 | Negative | Anemia | 66 | M | CYC | 409 | NIP, TRBV20-1 | 84, 24 | CASSLEIAGGLIYNEQFF, CASSPGLAGGRETQYF | 27, 2 | 0.093 | 0.350 |
| CCF15 | Negative | Anemia, neutropenia | 60 | M | CSA, CYC, MDX | 4472 | TRBV19 | 97 | CASSIGIQPQHF | 83 | 0.686 | 0.791 |
| CCF16 | Negative | Asymptomatic | 59 | M | SCT, CYC | 1515 | TRBV27 | 30 | CASSSVGYEQYF | 40 | 0.173 | 0.478 |
| CCF17 | Negative | Anemia | 71 | F | MTX | 2536 | TRBV19 | 20 | CASRASGSDEQYF, CARSRDKGTDYGYTF | 10, 8 | 0.024 | 0.263 |
| CCF18 | Negative | Asymptomatic | 54 | M | None | 686 | TRBV27, TRVB4-3 | 12, 8 | CASSLGNYEQYF, CASSISQNTEAFF | 4, 7 | 0.026 | 0.319 |
AAD, age at diagnosis; CSA, cyclosporine-A; CYC, cyclophosphamide; F, female; FU, follow-up; M, male; MTX, methotrexate; SCT, stem cell transplant.
*At earliest sampling.
†Clonal diversity and Clonality assessed according to Clemente et al.9
In mutant patients, STAT3 clonal expansions and contractions paralleled those of TCR clones. In some illustrative cases, the TCR clonal burden of the dominant clone was higher than that of STAT3MT, suggesting that STAT3MT is not the ancestral event for clonal expansion (see patients CCF1, CCF2, CCF7, CCF8, and CCF9). Our findings imply that the STAT3 hit occurs as a secondary event within the preexpanded immunodominant clone. However, in other patients, the clonal burden of both the TCR and STAT3MT clone were similar (patients CCF3, CCF5, and CCF6), indicating that the STAT3MT may contribute to autonomous antigen-independent clonal expansion.
Serial samplings illustrated clonal dynamics in response to therapy. More than half of patients with T-LGLL were treated with 1 or more immunosuppressive therapy (IST) regimens, leading to hematologic response in 40% of those treated. Distinct patterns of clonal dynamics were seen after treatment and are illustrated as fish plots (Figure 2). Samples were not available at the first appearance of a clone, but the clonal expansion can be speculated based on samplings at other points. In some patients (4/18), both the STAT3MT (if present) and major TCR clone decreased in response to treatment (patients CCF1, CCF3, CCF5, and CCF10). In others (3/18), the clones persisted despite a hematologic response (patients CCF2, CCF4, and CCF17), suggesting major clones were functionally silenced in their ability to inhibit/destroy specific hematopoietic progenitors. We also observed a common phenomenon of TCR “clonotype switching” in many patients (6/18), wherein therapy contracts 1 major clonotype while another previously “minor” clonotype expands (patients CCF6, CCF11, CCF12, CCF14, CCF16, and CCF18), likely because of its relative therapy insensitivity. Interestingly, all newly emerging clones were STAT3WT (wild type), and half the patients with “switching” were resistant to IST therapy. Multiple clonotypes were present at initial sampling in a few patients without STAT3MT and persisted at the same rate in subsequent samplings, precluding identification of a truly immunodominant clonotype (4/18). It is thus possible that a small but highly pathogenic clone may have been missed in our initial analysis. Predictably, a stable or increasing clonal burden of both STAT3MT and VB CDR3 sequence was seen in nonresponders to IST (28%; patients CCF7, CCF9, CCF13, CCF15, and CCF17).
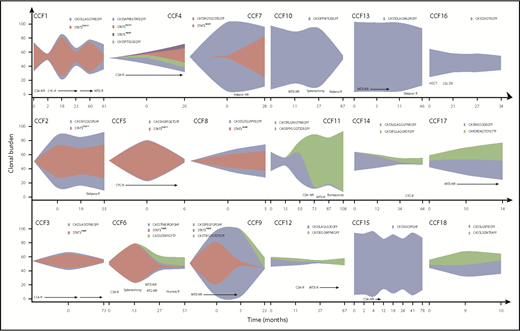
Clonal dynamics of STAT3 and TCR clonotypes throughout clinical course of patients with T-LGLL. Fish plots depicting the clonal expansions of 18 patients with T-LGLL. Sampling points are shown along the x-axis, and the clonal burden is shown on the y-axis. NR, no response; R, response.
Clonal dynamics of STAT3 and TCR clonotypes throughout clinical course of patients with T-LGLL. Fish plots depicting the clonal expansions of 18 patients with T-LGLL. Sampling points are shown along the x-axis, and the clonal burden is shown on the y-axis. NR, no response; R, response.
Discussion
Our results demonstrate that STAT3MT can arise either within an already preexpanded clonotype, or simultaneously with the clonal expansion of the immunodominant TCR Vβ clonotype. This leads us to believe that the STAT3MT mutation is selected among the originally triggered (responding) T-cell clones, and thus make the response more autonomous by either more persistence or less defendant on accessory signaling while not making the clone totally autonomous. The dynamics of both the STAT3MT and the TCR Vβ clonotype can be assessed over the course of the disease and in response to treatment regimens, and may demonstrate additional clinical utility when applied to larger prospective clinical trials (supplemental Figure 2). The difficulty in finding a direct correlation between response to a specific IST and a decrease in TCR Vβ clonal burden and STAT3MT may be a result of variability in time frames between samplings, IST regimens used, and the quality of response. Some large clonotypes may also be asymptomatic/not pathogenic, and thus may be overshadowing signals from smaller truly pathogenic clonotypes. Associations of remission with elimination of immunodominant clonotypes remain unclear. Our results suggest that pathogenic clones can be contracted to manageable clonal burdens and/or functionally silenced/tolerated. Thus, clonal elimination may not be needed for a complete clinical response.
The data on the technologies platform is publicly available at: https://clients.adaptivebiotech.com/pub/kerr-2019-bloodadvances (DOI: 10.21417/CK022019).
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute grants R01HL118281, R01HL123904, R01HL132071, and R35HL135795. S.M. was funded by the European Research Council (M-IMM Project) and the Academy of Finland.
Authorship
Contribution: C.M.K. performed sequencing experiments, collected/analyzed data, and wrote the manuscript; M.J.C collected data, advised on experiments, contributed to study design, and edited the manuscript; P.W.C. assisted with sequencing experiments; B.P., Y.N., V.A., and V.V. contributed to study design; T.R. reviewed the manuscript; A.E.L. and M.A.S. collected patient samples and clinical information; S.M. performed research; and J.P.M. designed and conceptualized the overall research and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jaroslaw P. Maciejewski, Taussig Cancer Center, Cleveland Clinic, 9500 Euclid Ave, Cleveland, OH 44195; e-mail: maciejj@ccf.org.

