

TO THE EDITOR:
Historically, metabolic studies in platelets have primarily investigated events occurring during ex vivo platelet storage, less so the consequences of metabolic alterations on platelet homeostasis and physiologic function. The importance of such studies is emphasized by the recent finding of platelet mitochondrial dysfunction in type 2 diabetes, sickle cell disease, and sepsis.1-7 These early investigations used radiometric methods with labeled glucose to study glycolysis and oxidative phosphorylation in platelets.8,9 Recently, these techniques have been supplanted by more sophisticated investigative tools that allow more rapid analysis of small volume samples, including high-resolution respirometry10,11 and Seahorse extracellular flux analysis.12-14 Glycolysis and oxidative phosphorylation, as well as glutaminolysis and fatty acid β oxidation, all function to support the metabolic demand in platelets.10,14-16 The metabolic flexibility of the platelet has been emphasized by investigations demonstrating that activated platelets exhibit a glycolytic phenotype while preserving mitochondrial function and can easily switch between glucose and fatty acid catabolism to support activation.12
The recently published study by A. K. Chauhan’s group further advances our understanding of the understudied field of platelet metabolism.13 In this paper, the authors demonstrate that dichloroacetate, an inhibitor of pyruvate dehydrogenase kinases, alters platelet metabolism and function.13 Finding salutary antiaggregatory and antithrombotic effects of dichloroacetate, the authors propose targeting of the platelet metabolic response as a novel antithrombotic approach. However, there are few points of clarification we would like to add, which we believe will be of great benefit for the platelet and mitochondria research community in their exploration of this new therapeutic avenue.13
As pointed out by the investigators in this study, the extracellular acidification rate (ECAR) is a commonly used indirect measure of glycolytic rate. Key to the interpretation of this indirect measure is the test of the cell’s utilization of glucose in the presence of a series of well-defined inhibitors.12,17 Dissection of the cause of acidification is necessary, as the relationship between ECAR and glycolysis is complicated by the existence of multiple acidification mechanisms, both mitochondrial and nonmitochondrial.17 As illustrated in Figure 1, CO2 is generated within the mitochondrial matrix by the pyruvate dehydrogenase complex and through the Krebs cycle. Upon diffusion from the cell, this mitochondrially generated CO2 is rapidly hydrated to H2CO3, which dissociates to bicarbonate ion and a proton at the physiological pH of the extracellular environment. Thus, conversion of 1 molecule of glucose to lactate (so called anaerobic glycolysis) yields 2 protons, whereas a complete oxidation of glucose to CO2 by mitochondrial mechanisms yields 6 protons. In addition, platelet mitochondria can also oxidize glutamine and fatty acids to produce substrates for the Krebs cycle.9,15,16,18-24 Thus, measurement of the glycolytic rate would require subtraction of acidification by mitochondrially derived CO2.
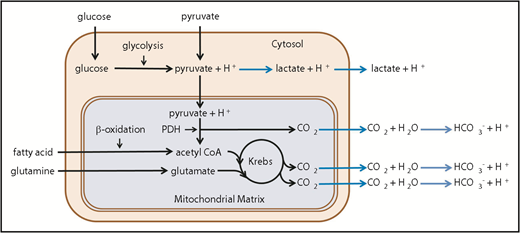
Summary of platelet catabolic pathways. Reactions leading to extracellular acidification caused by production of lactate and CO2 are shown with blue arrows. CoA, coenzyme A; Krebs, Krebs or tricarboxylic acid cycle; PDH, pyruvate dehydrogenase complex.
Summary of platelet catabolic pathways. Reactions leading to extracellular acidification caused by production of lactate and CO2 are shown with blue arrows. CoA, coenzyme A; Krebs, Krebs or tricarboxylic acid cycle; PDH, pyruvate dehydrogenase complex.
Another point we would like to address is the terminology used to describe cellular respiration, specifically the term aerobic glycolysis. Classically, cellular respiration is divided into 4 parts: glycolysis, pyruvate dehydrogenation, the Krebs cycle, and the electron transport chain coupled with chemiosmosis, with the last of these being the only oxygen-utilizing catabolic process. Pyruvate oxidation and the Krebs cycle are, however, dependent on oxidative phosphorylation and therefore would not occur in anaerobic conditions. Aerobic glycolysis, originally called the Warburg effect, is a phenomenon attributed mostly to cancer cells, which often rely primarily on the glycolytic part of glucose catabolism regardless of oxygenation.25-28 It is thought that the Warburg effect is a pathophysiologic adaptation of cancer cells to hypoxic conditions during early tumorigenesis. We propose that the term aerobic glycolysis be reserved for this unique setting and suggest that the term glycolysis (without the modifier) is sufficient to describe this aspect of glucose catabolism in platelets.
Adenosine triphosphate (ATP) plays a central role in the transfer of energy from its site of production to its site of utilization. Platelets, unlike many other cells, must abruptly transition from a “resting” state to an activated state with a similarly abrupt increase in energy consumption. The sudden increase in energy demand from the “burning” of the ATP resource during platelet activation has to be compensated if the newly activated platelet is to function within the hemostatic plug. Nayak et al have investigated a novel potential therapeutic avenue, namely preventing thrombus formation by altering the platelet’s catabolic response to activation.13 Glycolysis, although faster than oxidative phosphorylation, is not nearly as efficient in its ability to generate ATP, suggesting that an increase in oxidative phosphorylation should occur following platelet activation. Supporting this theoretical presumption, convulxin (CVX) stimulation increases both ECAR and oxygen consumption. This increase is inhibited by dasatinib, an inhibitor of the early steps of glycoprotein VI signaling.
To understand the mechanism driving this increase in oxygen utilization following CVX stimulation, we have measured the oxygen consumption rate and ECAR in the presence of various inhibitors of cyclooxygenase (acetyl salicylic acid) and lipoxygenase (2-(1-thienyl)ethyl 3,4-dihydroxybenzylidenecyanoacetate and 8,11,14-eicosatriynoic acid), none of which demonstrated an effect on metabolism in convulxin-stimulated platelets (Figure 2A-D). Preincubation of platelets with rotenone and antimycin A completely prevented this CVX-induced burst in oxygen consumption, indicating that this increased oxygen utilization is primarily of mitochondrial origin (Figure 2E-F), whereas the residual increase ECAR (Figure 2G-H) in the presence of these mitochondrial inhibitors is consistent with a compensatory increase in glycolysis (Figure 1). Distinction of the precise mechanisms of platelet metabolism will be crucial in the evaluation and development of antithrombotic therapies based on the novel therapeutic approach proposed by this study’s authors.

Glycoprotein VI–stimulated platelet respiration. Oxygen consumption (A-B,E-F) and extracellular acidification (C-D,G-H) in isolated human platelets. n ≥ 4; *P < .05 vs dimethyl sulfoxide. 2-TEDC, 2-(1-thienyl)ethyl 3,4-dihydroxybenzylidenecyanoacetate; 8,11,14-ETA, 8,11,14-eicosatriynoic acid; AA, antimycin A; ASA, acetyl salicylic acid; DMSO, dimethyl sulfoxide; OCR, oxygen consumption rate; PLT, platelet; Rot, rotenone.
Glycoprotein VI–stimulated platelet respiration. Oxygen consumption (A-B,E-F) and extracellular acidification (C-D,G-H) in isolated human platelets. n ≥ 4; *P < .05 vs dimethyl sulfoxide. 2-TEDC, 2-(1-thienyl)ethyl 3,4-dihydroxybenzylidenecyanoacetate; 8,11,14-ETA, 8,11,14-eicosatriynoic acid; AA, antimycin A; ASA, acetyl salicylic acid; DMSO, dimethyl sulfoxide; OCR, oxygen consumption rate; PLT, platelet; Rot, rotenone.
All studies were approved by the Institutional Review Board of Versiti Blood Research Institute.
Contribution: A.K. designed and performed the experiments and wrote the manuscript; and S.J. designed experiments and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Andaleb Kholmukhamedov, Versiti Blood Research Institute, 8727 W Watertown Plank Rd, Milwaukee, WI 53226, e-mail: andaleb.kholmukhamedov@versiti.org.

