

Key Points
Familial HLH can present as chronic isolated neuroinflammation.
CNS-isolated HLH responds to hematopoietic cell transplantation.
Introduction
Profound systemic inflammation traditionally characterizes hemophagocytic lymphohistiocytosis (HLH).1,2 Central nervous system (CNS) manifestations are common in HLH,3,4 but rarely precede systemic findings. In familial HLH, inherited mutations, most commonly in Prf1 and Unc13d, decrease lymphocyte cytotoxic function.5 Prf1 mutations obviate perforin production. Unc13d mutations prevent cell surface mobilization of CD107a and secretion of perforin and granzyme B.6 Case reports described Prf1 mutation patients with isolated CNS inflammation before treatment,4,7-10 but none suggested familial HLH can result in chronic, isolated neuroinflammation. The most recent international HLH treatment trial used etoposide, cyclosporine, and systemic steroids,1,2 but consensus regarding treatment of CNS involvement remains challenging.11
We describe 4 patients with chronic CNS-restricted familial HLH (CNS-HLH) lacking evidence of systemic inflammation. All achieved initial disease remission without requiring etoposide or cyclosporine, and tolerated consolidative allogeneic hematopoietic cell transplantation (HCT). Three patients achieved long-term disease remission and underlying molecular defect correction following first allogeneic HCT. The fourth relapsed following first HCT after not achieving molecular defect correction and required a second allogeneic HCT to achieve remission.
Methods
Patients and medical record review
The Boston Children’s Hospital Institutional Review Board approved this study. Clinical information came from the medical record.
HLH testing
Cincinnati Children’s Hospital Laboratories performed Prf1 and Unc13d gene sequencing, HLH gene panel next-generation sequencing, perforin staining, and CD107a mobilization.
CNS inflammation remission induction
Intrathecal methotrexate (12 mg) and hydrocortisone (15 mg) were administered every 2 weeks, and discontinued upon HCT conditioning.1 Systemic dexamethasone was dosed starting at 10 mg/m2 per day, weaned by 50% every other week until reaching 1.25 mg/m2 per day,1 and continued on this dose until weaning after HCT.
Bone marrow HCT procedure
Reduced-intensity conditioning consisted of alemtuzumab (0.2 mg/kg per day for 5 days starting 14 days before HCT12 or for 3 days starting 8 days before HCT13 ), fludarabine (30 mg/m2 per day for 5 days starting 8 days before HCT12 or 6 days starting 8 days before HCT13 ), and either melphalan (140 mg/m2 once 3 days before HCT12 ) or busulfan (0.8 mg/kg every 6 hours for 4 days starting 5 days before HCT,13 with a goal total area under the curve of 10 980-15 860 μmol × min per liter). Acute graft-versus-host disease (GVHD) prophylaxis included cyclosporine (troughs 250-350 ng/mL) and mycophenolate mofetil 15 mg/kg per day. The American Red Cross performed donor chimerism using short tandem repeat polymerase chain reaction.
Results and discussion
Four female patients with idiopathic neuroinflammation were ultimately diagnosed with CNS-HLH (baseline characteristics in Figure 1). Three patients (1, 2, and 4) experienced symptoms refractory to multiple therapies, with duration ranging from 2 to 6 years. The remaining patient (3) had asymptomatic radiologic neuroinflammation upon screening as a prospective sibling bone marrow donor for patient 2. Patients 1 and 4 suffered hemorrhagic strokes as inflammatory sequelae. Brain biopsy for patients 1, 2, and 4 demonstrated perivascular lymphocytic infiltration without hemophagocytosis.14 All patients had contrast-enhancing brain magnetic resonance imaging (MRI) lesions, which improved with high-dose corticosteroids, but ultimately could not be tolerated as chronic therapy. None of the patients experienced systemic inflammation (systemic HLH testing listed in supplemental Table 1), although it is unclear if prior therapy prevented such development. Cerebrospinal fluid (CSF) immunophenotyping and soluble biomarker analysis15 revealed innate immune activation in patient 1, leading to Prf1 gene sequencing. Imaging and biopsy similarities to patient 1 led to HLH gene panel sequencing for the remaining symptomatic patients.
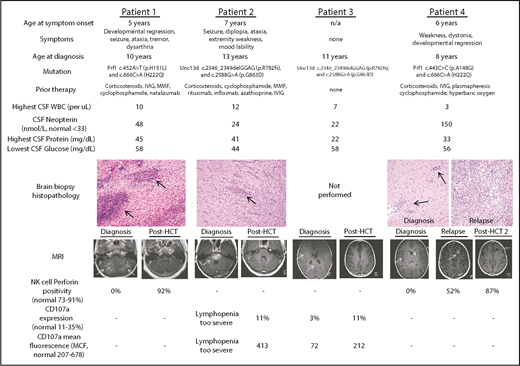
Baseline and posttreatment CNS-HLH patient characteristics. Baseline characteristics for all patients in this study, including age at onset of symptoms, nature of clinical symptoms, age at eventual diagnosis, familial HLH mutation, therapy received before CNS-HLH diagnosis, CSF assessment, and hematoxylin and eosin–stained brain biopsy histopathology (20× magnification). Black arrows indicate perivascular lymphocytic infiltrates. Also displayed are T1 MRI scans and NK-cell immunologic tests at diagnosis and after HCT. White arrows indicate contrast-enhancing MRI lesions. -, NK-cell testing not indicated for the patient’s specific mutation. HCT, hematopoietic cell transplantation; IVIG, intravenous immune globulin; MMF, mycophenolate mofetil; NK, natural killer; WBC, white blood cell.
Baseline and posttreatment CNS-HLH patient characteristics. Baseline characteristics for all patients in this study, including age at onset of symptoms, nature of clinical symptoms, age at eventual diagnosis, familial HLH mutation, therapy received before CNS-HLH diagnosis, CSF assessment, and hematoxylin and eosin–stained brain biopsy histopathology (20× magnification). Black arrows indicate perivascular lymphocytic infiltrates. Also displayed are T1 MRI scans and NK-cell immunologic tests at diagnosis and after HCT. White arrows indicate contrast-enhancing MRI lesions. -, NK-cell testing not indicated for the patient’s specific mutation. HCT, hematopoietic cell transplantation; IVIG, intravenous immune globulin; MMF, mycophenolate mofetil; NK, natural killer; WBC, white blood cell.
For remission induction, all patients received systemic dexamethasone.1 Patients 1 and 4 also received intrathecal methotrexate and hydrocortisone.1 No treatment complications were observed, and etoposide and cyclosporine were excluded during remission induction. Patients 1 through 3 received unrelated donor bone marrow. For initial transplant, patient 4 received bone marrow from a healthy HLA-matched sibling who inherited the H222Q Prf1 mutation only. All patients experienced acute, limited toxicities consistent with reduced-intensity conditioning and the ensuing neutropenic period. Table 1 details the HCT process and patient-specific toxicities. No patients experienced acute or chronic GVHD. Patient 4 underwent first HCT in the intensive care unit because of requiring multiple continuous infusions to control dystonia and spasms; this patient also experienced delayed platelet engraftment attributed to cytomegalovirus and adenovirus viremia. All patients achieved stable whole blood and T-cell donor chimerism of at least 20% and 23%, respectively.
HCT outcomes for CNS-HLH patients
| Patient | 1 | 2 | 3 | 4 (first SCT) | 4 (second SCT) |
|---|---|---|---|---|---|
| Bone marrow donor source | HLA-B, HLA-C mismatched unrelated | HLA-matched unrelated | HLA-matched unrelated | HLA-matched sibling | HLA-matched unrelated |
| Conditioning* | Reduced-intensity alemtuzumab (−14), fludarabine (5 d), melphalan | Reduced-intensity alemtuzumab (−14), fludarabine (5 d), melphalan | Reduced-intensity alemtuzumab (−14), fludarabine (5 d), melphalan | Reduced-intensity alemtuzumab (−14), fludarabine (5 d), melphalan | Reduced-intensity alemtuzumab (−8), fludarabine (6 d), busulfan |
| Total marrow cell, dose/kg | 3.32e+08 | 2.82e+08 | 5.29e+08 | 5.93e+08 | 5.30e+08 |
| Duration to myeloid engraftment, d† | 14 | 13 | 13 | 13 | 16 |
| Duration to platelet engraftment, d† | 12 | 19 | 17 | 144 | 14 |
| Patient-specific toxicities | Pericardial effusion, posttransplant microangiopathy (not requiring eculizumab) | Disseminated adenovirus infection, parainfluenza virus respiratory infection, Clostridium difficile colitis | C difficile colitis | Adenovirus viremia cytomegalovirus viremia, Staphylococcus aureus and α-hemolytic Streptococcus bacteremia, recurrent C difficile colitis | CMV reactivation, coagulase negative Staphylococcal bacteremia |
| Day +100 chimerism | CD3 and WB >97% donor | CD3 13%, WB 95% donor | CD3 26%, WB 52% donor | CD3 33%, WB 79% donor | CD3 69%, WB >97% donor |
| Most recent chimerism | 37 mo: CD3, WB, CD33/66 >97% | 27 mo: CD3 91%, WB 91%, CD33/66, CD56 >97% | 23 mo: CD3 23%, WB 20%, CD33/66 9%, CD56 22% | At relapse: CD3 46%, WB 47%, CD33/66 51%, CD56 50% | 14 mo: CD3 91%, WB >97%, CD33/66, CD56 >97%, |
| NK-cell perforin positivity (normal, 73%-91%) | 37 mo: 93% | NP | NP | At relapse: 52% | 3 mo: 87% |
| CD107a expression (normal, 11%-35%) | NP | 20 mo: 11% | 17 mo: 11% | At relapse: 11% | NP |
| CD107a MCF (normal, 206-678 MCF) | NP | 20 mo: 413 | 17 mo: 212 | At relapse: 209 | NP |
| Most recent MRI monitoring, mo | 30 | 23 | 17 | 13 | 9 |
| Duration of follow-up post-BMT, mo | 39 | 32 | 28 | Relapse: 13 | 15 |
| Patient | 1 | 2 | 3 | 4 (first SCT) | 4 (second SCT) |
|---|---|---|---|---|---|
| Bone marrow donor source | HLA-B, HLA-C mismatched unrelated | HLA-matched unrelated | HLA-matched unrelated | HLA-matched sibling | HLA-matched unrelated |
| Conditioning* | Reduced-intensity alemtuzumab (−14), fludarabine (5 d), melphalan | Reduced-intensity alemtuzumab (−14), fludarabine (5 d), melphalan | Reduced-intensity alemtuzumab (−14), fludarabine (5 d), melphalan | Reduced-intensity alemtuzumab (−14), fludarabine (5 d), melphalan | Reduced-intensity alemtuzumab (−8), fludarabine (6 d), busulfan |
| Total marrow cell, dose/kg | 3.32e+08 | 2.82e+08 | 5.29e+08 | 5.93e+08 | 5.30e+08 |
| Duration to myeloid engraftment, d† | 14 | 13 | 13 | 13 | 16 |
| Duration to platelet engraftment, d† | 12 | 19 | 17 | 144 | 14 |
| Patient-specific toxicities | Pericardial effusion, posttransplant microangiopathy (not requiring eculizumab) | Disseminated adenovirus infection, parainfluenza virus respiratory infection, Clostridium difficile colitis | C difficile colitis | Adenovirus viremia cytomegalovirus viremia, Staphylococcus aureus and α-hemolytic Streptococcus bacteremia, recurrent C difficile colitis | CMV reactivation, coagulase negative Staphylococcal bacteremia |
| Day +100 chimerism | CD3 and WB >97% donor | CD3 13%, WB 95% donor | CD3 26%, WB 52% donor | CD3 33%, WB 79% donor | CD3 69%, WB >97% donor |
| Most recent chimerism | 37 mo: CD3, WB, CD33/66 >97% | 27 mo: CD3 91%, WB 91%, CD33/66, CD56 >97% | 23 mo: CD3 23%, WB 20%, CD33/66 9%, CD56 22% | At relapse: CD3 46%, WB 47%, CD33/66 51%, CD56 50% | 14 mo: CD3 91%, WB >97%, CD33/66, CD56 >97%, |
| NK-cell perforin positivity (normal, 73%-91%) | 37 mo: 93% | NP | NP | At relapse: 52% | 3 mo: 87% |
| CD107a expression (normal, 11%-35%) | NP | 20 mo: 11% | 17 mo: 11% | At relapse: 11% | NP |
| CD107a MCF (normal, 206-678 MCF) | NP | 20 mo: 413 | 17 mo: 212 | At relapse: 209 | NP |
| Most recent MRI monitoring, mo | 30 | 23 | 17 | 13 | 9 |
| Duration of follow-up post-BMT, mo | 39 | 32 | 28 | Relapse: 13 | 15 |
Parentheses indicate number of days before HCT for start of alemtuzumab administration or total number of days of fludarabine administration.
Myeloid engraftment defined as absolute neutrophil count stably >0.5 × 109/L; platelet engraftment defined as platelet count stably >50 × 109/L.
CD3, T-cell donor chimerism; CD33/66, myeloid donor chimerism; CD56, NK-cell donor chimerism; MCF, mean channel fluorescence; NP, not performed; SCT, stem cell transplantation; WB, whole blood donor chimerism.
Following HCT, patients 1 through 3 corrected prior NK-cell perforin deficiency or CD107a mobilization defects. Abnormal MRI contrast enhancement resolved for all 3 (Figure 1). No further neurologic deficits developed, and patients 1 and 2 experienced improvements in weakness, ataxia, and diplopia, and markedly decreased seizure frequency. All 3 discontinued immunosuppressive corticosteroid by day 96 post-HCT, and have been followed for 28 to 39 months post-HCT.
Patient 4 initially resolved abnormal MRI enhancement and improved clinically, but at 13 months developed seizures and a new enhancing lesion (Figure 1). Extensive testing excluded infection and posttransplant lymphoproliferative disorder; biopsy demonstrated a destructive lymphocytic infiltrate (Figure 1).14 She was diagnosed with relapsed CNS-HLH. She met no systemic HLH criteria (supplemental Table 1), but had low perforin positivity. Whole blood, T-cell, and NK-cell donor chimerism was >46%. Upon patient 4’s relapse, immunologic testing of the sibling donor revealed low NK-cell perforin positivity (64%) and CD107a mobilization (6% of cells with expression; mean fluorescence, 183). Because only Prf1 sequencing was obtained before HCT, broader genetic testing is awaiting donor consent. Patient 4 repeated remission induction with dexamethasone before undergoing second HCT with an unrelated donor and an alternative reduced-intensity conditioning regimen substituting busulfan for melphalan and increasing total fludarabine dose. Following the second HCT, patient 4 has normalized NK-cell defects (Figure 1), higher unrelated donor chimerism, and no abnormal MRI enhancement. No additional neurologic symptoms or seizures have developed 15 months following the second HCT.
In this report, we identify a novel spectrum of CNS-isolated disease manifestations for an inherited hematologic disorder previously thought to primarily induce overwhelming systemic inflammation. The subtle onset of CNS-HLH can result in progressive, devastating neurologic complications, as evidenced in patients 1 and 4. A high index of suspicion for CNS-HLH must be maintained for all patients with idiopathic neuroinflammation, with urgent hematologist referral should genetic testing return positive for familial HLH.
Treatment with CNS-directed HLH therapy immediately upon confirmation of diagnosis may prevent symptom development or neurologic sequelae, as evidenced by patient 3. For remission induction of CNS-HLH, we found systemic dexamethasone combined with intrathecal methotrexate and hydrocortisone to be effective, without requiring etoposide or cyclosporine. Deferring the use of nephrotoxic agents is key in preserving renal function before HCT.
HCT using reduced-intensity conditioning regimens was tolerated without engraftment failure or acute GVHD. Three of 4 patients achieved disease control following first HCT. All achieved donor chimerism >20% reported as protective against systemic HLH recurrence.16 Patient 4 relapsed in the setting of persistently decreased perforin expression despite adequate donor chimerism. Further mechanistic investigation is required to determine if the lower NK-cell perforin positivity observed in genetic carriers17 is inadequate for disease control in CNS-HLH, or if higher donor chimerism is required. These findings underscore the importance of screening related stem cell donors with immunologic testing and broader genetic testing should NK-cell abnormalities be discovered.
Although all patients in this cohort achieved disease control, longer term follow-up is required to assess response durability given reports of CNS-isolated relapses following HCT for systemic HLH.18,19 Follow-up will also help clarify optimal post-HCT monitoring protocols, including MRI, CSF, donor chimerism, and NK-cell testing. Nevertheless, our results suggest HCT is an effective treatment of CNS-restricted HLH.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the Stem Cell Transplant, Neurology, Rheumatology, and Medical Intensive Care clinical services at Boston Children’s Hospital for providing clinical care, as well as the Undiagnosed Diseases Program at the National Institutes of Health for assistance with diagnostic testing. The authors acknowledge Biogen (Cambridge, MA) for compassionate use of natalizumab in patient 1 before her central nervous system–restricted familial hemophagocytic lymphohistiocytosis diagnosis.
H.L. is funded by an American Society of Hematology Research Training Award for Fellows grant.
Authorship
Contribution: All authors participated in the characterization of the disease process and treatment outcomes; H.L. and C.N.D. wrote the manuscript; and all authors edited the manuscript.
Conflict-of-interest disclosure: L.A.B. receives research support from Biogen Idec for clinical trial participation. B.B. has received patent royalty payments from the National Institutes of Health (NIH) for patents related to daclizumab therapy of multiple sclerosis. M.P.G. receives research funding from Biogen Idec and Novartis for clinical trial site participation and clinical research support from Pfizer, NIH, and the National Multiple Sclerosis Society. C.N.D. is a consultant for Bluebird Bio, Magenta Therapeutics, and AbGenomics. The remaining authors declare no competing financial interest.
Correspondence: Christine N. Duncan, Dana-Farber/Boston Children’s Cancer and Blood Disorders Center, 450 Brookline Ave, Boston, MA 02215; e-mail: christine_duncan@dfci.harvard.edu; and Mark P. Gorman, Department of Neurology, Boston Children’s Hospital, 300 Longwood Ave, Boston, MA 02115; e-mail: mark.gorman@childrens.harvard.edu.

