

Key Points
Ibrutinib but not zanubrutinib induces shedding of GPIb-IX complex in an ADAM17-dependent manner; GPIX has not been shown previously.
Ibrutinib, but not zanubrutinib, induces shedding of integrin αIIbβ3 by an unknown sheddase.

Abstract
The Bruton’s tyrosine kinase (Btk) inhibitor ibrutinib has proven to be efficacious in the treatment of B-cell chronic lymphocytic leukemia (B-CLL) and related diseases. However, a major adverse side effect of ibrutinib is bleeding, including major hemorrhages. The bleeding associated with ibrutinib use is thought to be due to a combination of on-target irreversible Btk inhibition, as well as off-target inhibition of other kinases, including EGFR, ITK, JAK3, and Tec kinase. In this study, we investigated the effects of ibrutinib vs zanubrutinib (a more selective Btk inhibitor) on platelet activation, glycoprotein expression, and thrombus formation. Ibrutinib, but not zanubrutinib, induced a time- and dose-dependent shedding of GPIb-IX complex and integrin αIIbβ3, but not of GPVI and GPV, from the platelet surface. The shedding of GPIbα and GPIX was blocked by GM6001 and TAPI-2, an ADAM17 inhibitor but not ADAM10 inhibitor. Ibrutinib but not zanubrutinib treatment of human platelets increased ADAM17 activation. Pretreatment of C57BL/6 mice with ibrutinib (10 mg/kg), but not zanubrutinib (10 mg/kg), inhibited ex vivo and in vivo thrombus growth over time. Platelets from ibrutinib-treated patients with CLL showed reduced GPIb-IX complex and integrin αIIbβ3 surface expression and reduced ex vivo thrombus formation under arterial flow, which was not observed in zanubrutinib-treated patients. In mice, ibrutinib, but not zanubrutinib, led to increased soluble GPIbα and soluble αIIb levels in plasma. These data demonstrate that ibrutinib induces shedding of GPIbα and GPIX by an ADAM17-dependent mechanism and integrin αIIbβ3 by an unknown sheddase, and this process occurs in vivo to regulate thrombus formation.
Introduction
Ibrutinib is an irreversible inhibitor of Bruton’s tyrosine kinase (Brk), a member of the Tec family that is essential in B-cell antigen receptor signaling.1-3 Ibrutinib is approved for treatment of patients with chronic lymphocytic leukemia (CLL), mantle cell lymphoma, and Waldenström macroglobulinemia.3-8 Ibrutinib is also improved for marginal zone lymphoma and graft-versus-host disease in the United States.3-8 Although ibrutinib is generally well tolerated, clinical studies have reported grade ≥3 bleeding events, including hematomas, hematuria, and gastrointestinal bleeding in 5% to 6% of patients.9 This bleeding risk is increased by concurrent therapy with antiplatelet agents or anticoagulants, including warfarin and direct oral anticoagulants.9 This is a particular problem in the elderly, who often have concurrent antithrombotic therapy and who are underrepresented or excluded from clinical trials.9
Btk plays an essential role in the GPIb and GPVI signaling pathways, which mediate adhesion to von Willebrand factor (VWF) and collagen, particularly in high shear rates in vivo.10-14 Patients with X-linked agammaglobulinemia do not have bleeding problems, despite the absence of functional Btk.15 This is consistent with the observation that the second-generation Btk inhibitors acalabrutinib and zanubrutinib have been reported to have lower bleeding rates compared with ibrutinib in clinical trials. This finding suggests that off-target effects of ibrutinib may contribute to bleeding.16-18 Zanubrutinib has a more specific targeted binding profile than ibrutinib with less off-target effects on enzymes, including tyrosine kinase expressed in hematopoietic carcinoma, human epidermal growth factor receptor 2, Janus kinase 3 (JAK3), epidermal growth factor receptor, and interleukin 2-inducible T-cell kinase.19 Previous studies have demonstrated that ibrutinib affects collagen and 4 VWF-dependent platelet functions but not G-protein coupling–dependent functions.12 In addition, ibrutinib inhibits platelet integrin αlIbβ3 outside-in signaling and thrombus stability, but not adhesion to collagen.20 These findings suggest that ibrutinib therapy is associated with complex effects on multiple platelet signaling pathways that contribute to bleeding events.
Platelet receptor internalization and/or shedding is thought to be important in clearing aged platelets from the circulation and regulates surface expression of a range of glycoproteins, including GPIb-IX-V complex and GPVI.21 Receptor shedding is induced by agonists and ADAM family members. GPIbα is exclusively shed by agonist-induced activation of ADAM17 protease, resulting in generation of soluble GPIbα fragments. In contrast, collagen GPVI receptor is normally shed by agonist-induced activation of ADAM10 protease.21,22
In the present study, we investigated the effects of ibrutinib compared with a second-generation Btk inhibitor, zanubrutinib, on platelet function, glycoprotein expression, and thrombus formation in blood flow, by using platelets from C57BL/6 mice, healthy donors, and Btk inhibitor–treated patients with CLL. Ibrutinib-treated but not zanubrutinib-treated blood samples from healthy donors or Btk inhibitor-treated patients with B-CLL, demonstrated reduced thrombus formation on immobilized collagen, VWF, or fibrinogen in arterial flow conditions. Our experiments further showed that ibrutinib but not zanubrutinib treatment resulted in loss of GPIb-IX-V complex from the platelet surface in an ADAM17- but not ADAM10-dependent manner. This reduction was observed for GPIb-IX-V complex expression but not GPVI expression on the platelets in healthy donors treated in vitro by ibrutinib and a proportion of chronically treated B-CLL patients. These data demonstrate that ibrutinib induces shedding of GPIbα surface receptor by an ADAM17-dependent mechanism and that the process occurs in vivo.
Methods
Materials are listed in the supplemental Information.
Drugs
BGB-3111 (zanubrutinib; 97.60% purity) and BGB-1672 (ibrutinib; 98.75% purity) were supplied by Beigene. Ibrutinib was also extracted from capsules (Johnson and Johnson, North Ryde, NSW, Australia). All data analyses in this paper are based on the purified forms of ibrutinib and zanubrutinib.
Animals
All animal experiments and care were approved by the Royal Melbourne Institute of Technology (RMIT) University Animal Ethics Committee (AEC1628). C57BL/6 mice were used at 6 to 10 weeks of age.
B-CLL patients
All human Btk-treated B-CLL blood samples and experiments were approved by the Peter MacCallum Cancer Centre Human Research Ethics Committee (HREC), Project 18/206. Patient samples were from clinical trials BGB-3111-AU-003 and NCT02343120. All 5 ibrutinib-treated patients received commercial drug.
Platelet preparation
Platelet rich plasma (PRP) and washed human or murine platelets were isolated, and platelet aggregation experiments were performed as previously described.23-25
Hematological parameter analysis
Full blood examination parameters were determined as previously described.23-25
Measurement of α- and dense-granule exocytosis
Release of α and dense granules was performed as previously described.23
Clot retraction assessment
Clot retraction was performed as previously described.24,26
Tail bleeding time
Tail bleeding time was determined as previously described.24
Flow cytometry.
Washed platelets (50 µL of 100 × 109/L) were preincubated with the PE-conjugated antibodies human CD42a (GPIX), anti-human CD41a (integrin αIIb), anti-human CD61 (integrin β3), anti-human CD42b (GPIbα), anti-human GPVI, or anti-human CD42c (GPIbβ); APC-conjugated anti-human CD42d (GPV) antibodies; or FITC-conjugated PAC-1 antibodies. The subsequent steps and analysis have been described.23-25
Adhesion under flow conditions.
A 6-channel µ-Slide VI, with dimensions (0.1 × 1.0 × 17 mm, H × W × L; Microslides; Ibidi, Martinsried, Germany), was precoated with 500 µg/mL type I fibrillar collagen (Nycomed, Linz, Austria), 50 µg/mL VWF, or 100 µg/mL fibrinogen for 2 hours at 37°C, followed by washing with Tyrode’s buffer. Whole blood from healthy donor and Btk-treated CLL patients was normalized to 200 × 109/L platelets and incubated with 0.05% (w/v) rhodamine 6G dye for 30 minutes at 37°C. Labeled platelets were then perfused through the collagen-coated microslide at an arterial shear flow rate of 1800 seconds−1 for 6 minutes. Nonadherent cells were washed away with Tyrode’s buffer. Thrombi formation was recorded in real time over the 6-minute time frame using z-stack analysis and deconvolution with 3-dimensional (3D) reconstructions, using Zeiss Axiovert M1 microscope with AxioCam MRm camera (Zeiss, Thornwood, NY). Thrombus area (in square micrometers), thrombus height (micrometers), and thrombus volume (cubic micrometers) were determined at 2, 4, and 6 time points.
Ex vivo thrombus formation under arterial flow
C57BL/6 mice were treated with ibrutinib (10 mg/kg), zanubrutinib (10 mg/kg), or dimethyl sulfoxide (DMSO) control for 2 hours. Citrated whole blood was obtained from C57BL/6 mice, and the platelets were fluorescently labeled with 0.05% (w/v) rhodamine 6G dye. After incubation at 37°C for 30 minutes, labeled platelets in whole blood were then perfused over a matrix of type I collagen–coated µ-Slide VI0.1 for 6 minutes at a shear stress rate of 1800 seconds−1. Thrombus formation was monitored as described above.
Ferric chloride–induced vascular injury of mesenteric arterioles
C57BL/6 mice (4-6 weeks old) were orally administered with the Btk inhibitors 10 mg/kg ibrutinib and 10 mg/kg zanubrutinib or DMSO-treated control by oral gavage. After 2 hours (peak drug concentration), Btk inhibitor–treated mice were anesthetized with a ketamine/xylazine (100:10 mg/kg) mixture intraperitoneally. Ferric chloride (FeCl3) injury and intravital microscopy were performed as previously described.23
ADAM17 activity.
Washed platelets (200 µL of 100 × 109/L) were placed into a 96-well white plate (Perkin Elmer, Melbourne, VIC, Australia) and pretreated with 20 µM fluorogenic TACE substrate (7-methoxycoumarin-4-yl) acetyl-P LAQAV-N-3-(2,4-dinitrophenyl)-l-2,3-diaminopropionyl-RSSSR-NH2) at 37°C for 20 minutes. Platelets were then incubated with vehicle control, type I collagen (20 µg/mL), ibrutinib (0.5 µM), or zanubrutinib (0.5 µM) for 40 minutes at 37°C. The cleavage of the TACE fluorogenic substrate was monitored with a Clariostar microplate reader (BMG Labtech, Mornington, VIC, Australia) set at 320/405 nm.
ELISA.
Soluble murine GPIbα (Cusabio, Houston, TX) and soluble murine αIIb levels (Cusabio) were measured by solid phase sandwich enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions. Plasma was isolated from citrated whole blood collected from 8-week-old C57BL/6 mice treated with 10 mg/kg ibrutinib, 10 mg/kg zanubrutinib, or sham control for 2 hours. The samples were centrifuged at 1000g for 10 minutes to obtain platelet-poor plasma. All ELISA assays were read on a Victor X3 multilabel plate reader at 450 nm wavelength (Perkin Elmer, Waltham, MA).
Statistical analysis
Statistical analysis was performing using Prism software, version 7 (GraphPad, San Diego, CA). Data sets were expressed as the mean ± standard error of the mean (SEM). Statistical significance for 2 data sets was determined with the unpaired Student t test. A value of P ≤ .05 was considered significant.
Results
Ibrutinib, but not zanubrutinib, reduces platelet adhesion to type I collagen, VWF, and fibrinogen under arterial flow conditions
Ibrutinib and zanubrutinib are specific Btk inhibitors that target the same cysteine residue on Btk to regulate ITAM-dependent pathways. To examine the effect of ibrutinib and zanubrutinib on thrombus formation under high shear flow conditions in vitro, we incubated human blood with ibrutinib or zanubrutinib (0.5 µM) and studied platelet adhesion to type I collagen, VWF, or fibrinogen over time. Vehicle- and zanubrutinib-treated platelets (0.5 µM) adhered and formed stable thrombi over a 6-minute period. In contrast, ibrutinib-treated platelets (0.5 µM) dramatically reduced the number of adherent platelets and virtually abrogated thrombus formation, as shown by a significant reduction in thrombus volume on type I collagen matrix or VWF or fibrinogen over the 6-minute period (Figure 1). This indicated an antiplatelet mechanism induced by ibrutinib, but not by zanubrutinib, that directly interfered with platelet adhesion on collagen, VWF, and fibrinogen under conditions of arterial shear.
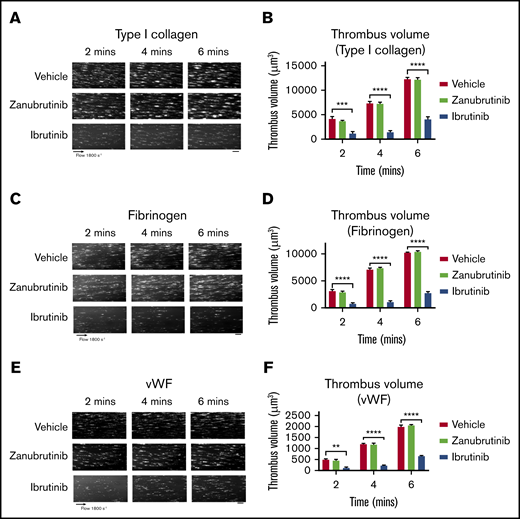
The effect of ibrutinib and zanubrutinib on in vitro human thrombus formation and growth. Fluorescently labeled whole human blood was either untreated or treated with 0.5 µM ibrutinib or 0.5 µM zanubrutinib at 37°C for 30 minutes before perfusion over 500 µg/mL type I collagen, 50 µg/mL VWF, or 100 µg/mL fibrinogen, under arterial flow conditions at a shear rate of 1800 seconds−1. Z-stack images were captured over a 6-minute period with a digital AxioCam MRm camera (Zeiss) and analyzed with Zeiss Axiovision Rel 4.6 software. 3D deconvolved reconstructions of the thrombi formed were analyzed for surface coverage of platelet aggregates (in square micrometers), thrombus height (in micrometers), and thrombus volume (in cubic micrometers). (A,C,E) Representative images of thrombus formation over type I collagen, VWF, or fibrinogen, in arterial flow conditions, in human whole blood treated with 0.5 µM ibrutinib, 0.5 µM zanubrutinib, or vehicle control over 6 minutes. Scale bars represent 20 μm. (B,D,F) Thrombus volume over time was calculated from thrombus area × thrombus height for each Btk inhibitor. Results are cumulative data from 4 independent experiments and are presented as the mean ± SEM by unpaired Student t test. **P ≤ .01, ***P ≤ .001, ****P ≤ .0001.
The effect of ibrutinib and zanubrutinib on in vitro human thrombus formation and growth. Fluorescently labeled whole human blood was either untreated or treated with 0.5 µM ibrutinib or 0.5 µM zanubrutinib at 37°C for 30 minutes before perfusion over 500 µg/mL type I collagen, 50 µg/mL VWF, or 100 µg/mL fibrinogen, under arterial flow conditions at a shear rate of 1800 seconds−1. Z-stack images were captured over a 6-minute period with a digital AxioCam MRm camera (Zeiss) and analyzed with Zeiss Axiovision Rel 4.6 software. 3D deconvolved reconstructions of the thrombi formed were analyzed for surface coverage of platelet aggregates (in square micrometers), thrombus height (in micrometers), and thrombus volume (in cubic micrometers). (A,C,E) Representative images of thrombus formation over type I collagen, VWF, or fibrinogen, in arterial flow conditions, in human whole blood treated with 0.5 µM ibrutinib, 0.5 µM zanubrutinib, or vehicle control over 6 minutes. Scale bars represent 20 μm. (B,D,F) Thrombus volume over time was calculated from thrombus area × thrombus height for each Btk inhibitor. Results are cumulative data from 4 independent experiments and are presented as the mean ± SEM by unpaired Student t test. **P ≤ .01, ***P ≤ .001, ****P ≤ .0001.
Ibrutinib and zanubrutinib inhibited α- and dense-granule exocytosis mediated by GPVI or CLEC-2
When platelets are stimulated, the processes of platelet activation are enhanced by α-granule release (P-selectin expression) and dense granule production of ADP and serotonin that make platelet aggregation more stable. To investigate the effect of Btk inhibitors (ibrutinib and zanubrutinib) on α- or dense-granule release, platelets pretreated with vehicle DMSO-treated or Btk inhibitor were stimulated with agonists, including thrombin (0.25 U/mL), PAR-1 (1.25 µM), PAR-4 (25 µM), CRP-XL (0.25 µg/mL), and rhodocytin (0.7 µg/mL). The concentration dependence of ibrutinib and zanubrutinib (0.5-2.0 µM) on agonist-induced α- and dense-granule exocytosis was examined. P-selectin expression was measured by flow cytometry with an FITC-labeled anti-CD62P antibody, whereas dense granule exocytosis was determined by quinacrine release. Ibrutinib or zanubrutinib significantly inhibited rhodocytin (CLEC-2 ligand)- and CRP-XL (GPVI selective agonist)–induced P-selectin expression and dense granule release at different doses of drugs (0.5-2.0 µM) (Figure 2A-J). In contrast, there was no effect of ibrutinib or zanubrutinib observed with thrombin, PAR-1–, or PAR-4–induced P-selectin expression and dense granule exocytosis. Ibrutinib but not zanubrutinib also inhibited PAC-1 binding (Figure 2K). These data indicate that both drugs inhibit GPVI and CLEC-1 hemi (ITAM)-mediated signaling pathways that modulate α- and dense-granule exocytosis.

The effect of Btk inhibitors on thrombin-, PAR-1-, PAR-4-, CRP-XL- and rhodocytin-mediated α- and dense-granule exocytosis. Washed human platelets (100 × 109/L) were incubated with a range of doses (0.5-2.0 μm) of ibrutinib or zanubrutinib for 10 minutes at 37°C, and either unstimulated (resting) or stimulated by thrombin (0.25 U/mL) (A), PAR-1 (1.25 μM) (B), PAR-4 (25 μM) (C), rhodocytin (0.7 µg/mL) (D), or CRP (0.25 µg/mL) (E). The platelets were then labeled with the FITC-P-selectin CD62P monoclonal antibody. Flow cytometric analysis was used to determine platelet P-selectin exposure. Quinacrine-labeled platelets were untreated or treated with a range of doses (0.5-2.0 μM) of ibrutinib or zanubrutinib for 10 minutes at 37°C. Platelets then were either unstimulated (resting) or stimulated by selective agonist thrombin (0.25 U/mL) (F), PAR-1 (1.25 µM) (G), PAR-4 (50 µM) (H), rhodocytin 0.7 µg/mL (I), and CRP 0.25 µg/mL (J). Flow cytometry analysis was used to determine the release of agonist-induced dense granule. (K) PAC-1 binding was determined for vehicle, ibrutinib, or zanubrutinib treatment after stimulation of washed human platelets (100 × 109/L) with thrombin (0.25 U/mL), PAR-1 (1.25 µM), PAR-4 (25 µM), rhodocytin (0.7 µg/mL), and CRP (0.25 µg/mL). Statistical analysis was performed with the unpaired Student t test. Results are represented as mean fluorescence intensity (MFI) ± SEM from 3 independent experiments. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001.
The effect of Btk inhibitors on thrombin-, PAR-1-, PAR-4-, CRP-XL- and rhodocytin-mediated α- and dense-granule exocytosis. Washed human platelets (100 × 109/L) were incubated with a range of doses (0.5-2.0 μm) of ibrutinib or zanubrutinib for 10 minutes at 37°C, and either unstimulated (resting) or stimulated by thrombin (0.25 U/mL) (A), PAR-1 (1.25 μM) (B), PAR-4 (25 μM) (C), rhodocytin (0.7 µg/mL) (D), or CRP (0.25 µg/mL) (E). The platelets were then labeled with the FITC-P-selectin CD62P monoclonal antibody. Flow cytometric analysis was used to determine platelet P-selectin exposure. Quinacrine-labeled platelets were untreated or treated with a range of doses (0.5-2.0 μM) of ibrutinib or zanubrutinib for 10 minutes at 37°C. Platelets then were either unstimulated (resting) or stimulated by selective agonist thrombin (0.25 U/mL) (F), PAR-1 (1.25 µM) (G), PAR-4 (50 µM) (H), rhodocytin 0.7 µg/mL (I), and CRP 0.25 µg/mL (J). Flow cytometry analysis was used to determine the release of agonist-induced dense granule. (K) PAC-1 binding was determined for vehicle, ibrutinib, or zanubrutinib treatment after stimulation of washed human platelets (100 × 109/L) with thrombin (0.25 U/mL), PAR-1 (1.25 µM), PAR-4 (25 µM), rhodocytin (0.7 µg/mL), and CRP (0.25 µg/mL). Statistical analysis was performed with the unpaired Student t test. Results are represented as mean fluorescence intensity (MFI) ± SEM from 3 independent experiments. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .0001.
Ibrutinib and zanubrutinib differently affect agonist-induced platelet aggregation and clot retraction in vitro
We evaluated the effect of pretreatment of platelet-rich plasma (PRP; platelets normalized to 100 × 109/L) with a dose-dependent range of concentrations of ibrutinib or zanubrutinib (0.25-1.0 µM; levels similar to those seen in the clinic) compared with DMSO-treated normal healthy human platelets on agonist-induced platelet aggregation responses as measured with a 4-channel platelet aggregometer. Consistent with the known role of Btk in GPVI signaling, ibrutinib and zanubrutinib inhibited CRP-XL, 1.0 µg/mL induced platelet aggregation at all doses compared with vehicle control. Ibrutinib and zanubrutinib also inhibited collagen-induced platelet aggregation when tested at 1.0 µg/mL, but to a lesser extent with zanubrutinib. Ibrutinib but not zanubrutinib also inhibited ADP- and TRAP-induced platelet aggregation at all drug doses tested (Figure 3A-D). This finding is in agreement with those in a previous study.27
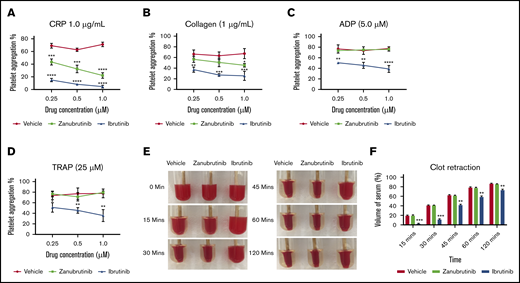
The effect of Btk inhibitors on agonist-induced platelet aggregation and clot retraction in vitro. (A-D) Human PRP was normalized to 100 × 109/L and incubated with a range of doses (0.25-1.0 µM) of ibrutinib, zanubrutinib, or vehicle control for 10 minutes at 37°C. Aggregation responses of PRP were determined after agonist stimulation by 1.0 μg/mL CRP (A), 1.0 μg/mL collagen (B), 5 μM ADP (C), and 25 μM TRAP (D). These results are represented as the mean ± SEM of 3 independent experiments. *P ≤ .05, ** P ≤ .01, *** P ≤ .005, **** P ≤ .001. Normalized PRP from a healthy human donor (100 × 109/L platelets) was left untreated or treated with 0.5 μM ibrutinib or 0.5 µM zanubrutinib for 5 minutes at 37°C. Clot formation was induced with 2.5 U/mL of thrombin and monitored over time at 37°C. (E) Representative images of clot retraction using PRP, untreated or treated with 0.5 μM ibrutinib or 0.5 µM zanubrutinib after addition of 2.5 U/mL of thrombin over time. (F) The remaining serum volume was measured and recorded after clot retraction at 15, 30, 45, 60, and 120 minutes. Data are representative of 4 independent experiments and are expressed as the mean ± SEM, by Student t test. **P ≤ .001, ***P ≤ .0001.
The effect of Btk inhibitors on agonist-induced platelet aggregation and clot retraction in vitro. (A-D) Human PRP was normalized to 100 × 109/L and incubated with a range of doses (0.25-1.0 µM) of ibrutinib, zanubrutinib, or vehicle control for 10 minutes at 37°C. Aggregation responses of PRP were determined after agonist stimulation by 1.0 μg/mL CRP (A), 1.0 μg/mL collagen (B), 5 μM ADP (C), and 25 μM TRAP (D). These results are represented as the mean ± SEM of 3 independent experiments. *P ≤ .05, ** P ≤ .01, *** P ≤ .005, **** P ≤ .001. Normalized PRP from a healthy human donor (100 × 109/L platelets) was left untreated or treated with 0.5 μM ibrutinib or 0.5 µM zanubrutinib for 5 minutes at 37°C. Clot formation was induced with 2.5 U/mL of thrombin and monitored over time at 37°C. (E) Representative images of clot retraction using PRP, untreated or treated with 0.5 μM ibrutinib or 0.5 µM zanubrutinib after addition of 2.5 U/mL of thrombin over time. (F) The remaining serum volume was measured and recorded after clot retraction at 15, 30, 45, 60, and 120 minutes. Data are representative of 4 independent experiments and are expressed as the mean ± SEM, by Student t test. **P ≤ .001, ***P ≤ .0001.
Integrin αIIbβ3 is crucial for normal platelet aggregation, platelet spreading, and clot retraction functionality. Therefore, we next examined whether there is any significant difference in the kinetics of the clot retraction process after Btk inhibitor treatment of PRP before addition of thrombin to induce clotting. Ibrutinib-treated PRP significantly delayed the kinetics of clot retraction over the 2-hour time frame, compared with zanubrutinib-treated PRP and vehicle control (Figure 3E-F).
Ibrutinib causes downregulation of GPIbα, GPIX, and integrin αIIbβ3, but not of GPVI, GPV, or GPIbβ from the platelet surface in a time- and dose-dependent manner
Several platelet receptors that are involved in the adhesion and aggregation process, including GPIb-IX-V complex, integrin αIIbβ3, and GPVI, are proteolytically downregulated in response to various agonists.24 Ibrutinib has been demonstrated to interfere with platelet adhesion on collagen but whether platelet glycoproteins are affected is not known. To investigate whether ibrutinib modulates the surface glycoprotein expression on platelets, we incubated whole blood with different concentrations of ibrutinib or zanubrutinib or vehicle control and measured the surface expression of the major glycoproteins on the platelet surface by flow cytometry. Ibrutinib but not zanubrutinib treatment of platelets caused a time- and dose-dependent downregulation of GPIbα, GPIX, and integrin αIIbβ3 (both αIIb and β3 subunits were reduced), but not GPVI, GPV, or GPIbβ (Figure 4A-K). This ibrutinib effect was observed with either a purified form of the drug or ibrutinib extracted from capsules that were given to patients with B-CLL (data not shown).
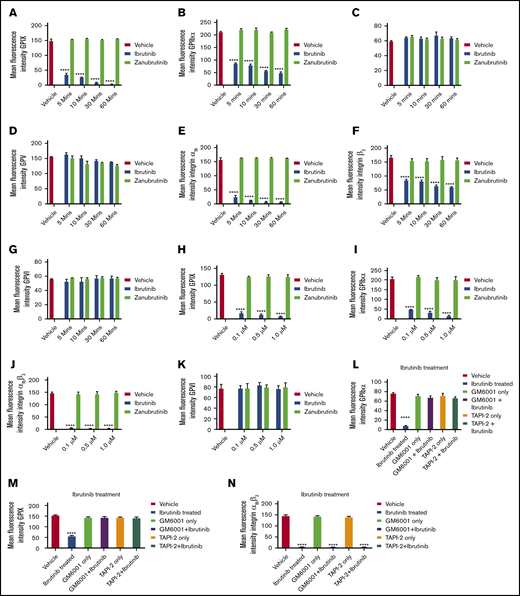
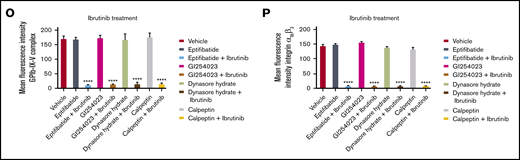
Ibrutinib treatment leads to loss of GPIbα, GPIX, and integrin αIIbβ3, but not GPVI, GPV, or GPIbβ from the platelet surface in a time- and dose-dependent manner. (A-K) Washed human platelets (100 × 109/L) were incubated to determine dose response (dose range, 0.1-1.0 µM) of ibrutinib or zanubrutinib or vehicle control for 5, 10, 30, and 60 minutes at room temperature (RT). The platelets were then labeled with anti-human CD42a (GPIX), anti-human CD42b (GPIbα), anti-human CD41a (integrin αIIb), anti-human CD61 (integrin β3), anti-human GPIbβ (CD42c), or anti-human GPVI PE-conjugated antibodies or anti-human GPV (CD42d) APC-conjugated antibodies. (L-N) Washed human platelets (100 × 109/L) were incubated with 100 µM GM6001 or 100 µM TAPI-2 for 2 hours at 37°C, followed by 0.5 µM ibrutinib treatment for 60 minutes at RT. The platelets were then labeled with anti-human CD42b, anti-human CD42a, or anti-human CD41a PE-conjugated antibodies. (O-P) Washed human platelets (100 × 109/L) were incubated with 100 µM GI254023, 100 µM calpeptin, 100 µM eptifibatide, or 100 µM Dynasore hydrate for 2 hours at 37°C followed by 0.5 µM ibrutinib treatment for 60 minutes at RT. The platelets were then labeled with anti-human CD42b or anti-human CD41a PE-conjugated antibodies. Flow cytometric analysis was used to determine the expression of GPIbα or integrin αIIbβ3. Statistical analysis was performed with the unpaired Student t test. Results are expressed as mean fluorescent intensity (MFI) ± SEM from 3 independent experiments. ****P ≤ .0001.
Ibrutinib treatment leads to loss of GPIbα, GPIX, and integrin αIIbβ3, but not GPVI, GPV, or GPIbβ from the platelet surface in a time- and dose-dependent manner. (A-K) Washed human platelets (100 × 109/L) were incubated to determine dose response (dose range, 0.1-1.0 µM) of ibrutinib or zanubrutinib or vehicle control for 5, 10, 30, and 60 minutes at room temperature (RT). The platelets were then labeled with anti-human CD42a (GPIX), anti-human CD42b (GPIbα), anti-human CD41a (integrin αIIb), anti-human CD61 (integrin β3), anti-human GPIbβ (CD42c), or anti-human GPVI PE-conjugated antibodies or anti-human GPV (CD42d) APC-conjugated antibodies. (L-N) Washed human platelets (100 × 109/L) were incubated with 100 µM GM6001 or 100 µM TAPI-2 for 2 hours at 37°C, followed by 0.5 µM ibrutinib treatment for 60 minutes at RT. The platelets were then labeled with anti-human CD42b, anti-human CD42a, or anti-human CD41a PE-conjugated antibodies. (O-P) Washed human platelets (100 × 109/L) were incubated with 100 µM GI254023, 100 µM calpeptin, 100 µM eptifibatide, or 100 µM Dynasore hydrate for 2 hours at 37°C followed by 0.5 µM ibrutinib treatment for 60 minutes at RT. The platelets were then labeled with anti-human CD42b or anti-human CD41a PE-conjugated antibodies. Flow cytometric analysis was used to determine the expression of GPIbα or integrin αIIbβ3. Statistical analysis was performed with the unpaired Student t test. Results are expressed as mean fluorescent intensity (MFI) ± SEM from 3 independent experiments. ****P ≤ .0001.
Ibrutinib causes downregulation of GPIbα and GPIX, but not of integrin αIIbβ3, in a metalloproteinase ADAM17-dependent manner
To characterize the mechanism underlying the ibrutinib-mediated loss of specific platelet glycoproteins and to identify the responsible metalloproteinase involved, we incubated whole human blood pretreated with GM6001, TAPI-2, or vehicle control before ibrutinib treatment. Pretreatment with the metalloproteinase inhibitor GM6001 or the ADAM17 inhibitor TAPI-2 before ibrutinib exposure blocked the loss of GPIbα and GPIX from the platelet surface (Figure 4L-N). In contrast, integrin αIIbβ3 was still lost from the platelet surface, even with pretreatment with GM6001 or TAPI-2. As ADAM17 is the exclusive metalloproteinase that mediates GPIbα and GPIX shedding from the platelet surface, these results suggest that ibrutinib mediates shedding of GPIbα, and GPIX occurs in an ADAM17-dependent manner.
To further characterize the mechanism underlying the ibrutinib-mediated loss of integrin αIIbβ3 and GPIbα and to identify the potential responsible protease involved, we incubated whole human blood pretreated with the ADAM10 inhibitor GI254023, the calpain inhibitor calpeptin, the dynamin inhibitor Dynasore hydrate, or the integrin αIIbβ3 antagonist eptifibatide. Pretreatment with the ADAM10 inhibitor (an ADAM family member in platelets), the calpain inhibitor (calpain is known to cleave β3 cytoplasmic domain in platelets), the dynamin inhibitor (to inhibit integrin αIIbβ3 internalization), or integrin αIIbβ3 antagonist (to inhibit integrin αIIbβ3 activation) did not block the loss of GPIbα or integrin αIIbβ3 from the platelet surface (Figure 4O-P). These results confirm that ibrutinib-mediated loss of GPIbα is specific to ADAM17 activity, and integrin αIIbβ3 is lost via an unknown sheddase in platelets.
Ibrutinib, but not zanubrutinib, inhibited in vitro and ex vivo murine thrombus formation on type I collagen during arterial flow, tail bleeding time, FeCl3-induced in vivo thrombus formation, and growth in C57BL/6 mesenteric arterioles
We next examined the effect of Btk inhibitors on murine thrombus formation of type I collagen during arterial flow, using whole blood derived from C57BL/6 mice. Citrated whole murine blood was treated with ibrutinib (0.5 µM) or zanubrutinib (0.5 µM), compared with vehicle control, for 30 minutes at 37°C. Whole blood was fluorescently labeled with rhodamine 6G before perfusion through a collagen-coated microcapillary tube flow chamber at an arterial shear rate of 1800 seconds−1. After 6 minutes of sample perfusion, thrombus images for Btk- vs vehicle-treated whole blood were recorded in real time and analyzed by 3D reconstructions. Ibrutinib inhibited thrombus growth, as shown by reduced surface coverage and thrombus volume over time (Figure 5A-B). In contrast, zanubrutinib had thrombus growth characteristics similar to those of the vehicle control on type I collagen during arterial flow over time.
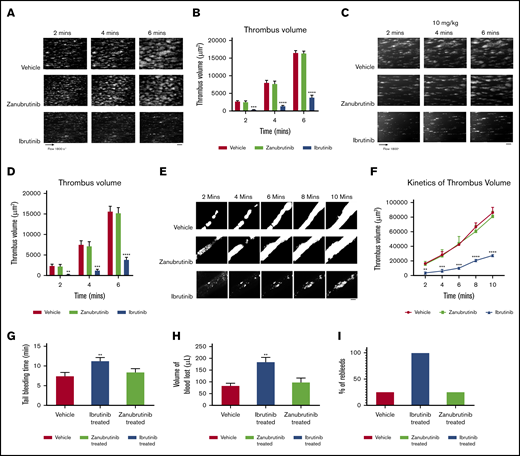
The effect of ibrutinib and zanubrutinib on in vitro, ex vivo, and in vivo thrombus formation and tail bleeding time. (A-B) Fluorescently labeled whole blood of C57BL/6 mice was untreated or treated with 0.5 μM of ibrutinib or 0.5 µM of zanubrutinib and was perfused over 500 μg/mL type I collagen under arterial flow conditions at a shear rate of 1800 seconds−1. Z-stack images were captured over a 6-minute duration with a digital AxioCam MRm camera (Zeiss) and analyzed with Zeiss Axiovision Rel 4.6 software. 3D deconvolved reconstructions of thrombi formed were analyzed for surface coverage of platelet aggregates (in square micrometers), thrombus height (in micrometers), and thrombus volume (in cubic micrometers). (A) Representative images of thrombi formation over type I collagen under arterial flow conditions for wild-type mouse whole blood treated with 0.5 μM of ibrutinib or 0.5 µM of zanubrutinib vs DMSO control over 6 minutes. Scale bar represents 20 μm. (B) Thrombus volume over time was calculated from thrombus area × thrombus height. Results are cumulative data from 4 independent experiments and presented as the mean ± SEM, by the unpaired Student t test. ***P ≤ .005, ****P ≤ .001. (C-D) Fluorescently labeled whole blood of C57BL/6 mice untreated or treated with 10 mg/kg ibrutinib or 10 mg/kg zanubrutinib was perfused over 500 μg/mL type I collagen under arterial flow conditions at a shear rate of 1800 seconds−1. Z-stack images were captured over a 6-minute duration with a digital AxioCam MRm camera (Zeiss) and analyzed with Zeiss Axiovision Rel 4.6 software. 3D deconvolved reconstructions of thrombi formed were analyzed for surface coverage of platelet aggregates, thrombus height, and thrombus volume. Scale bar represents 20 μm. (E) Representative images of thrombus formation over type I collagen under arterial flow conditions for wild-type mice treated with 10 mg/kg ibrutinib or 10 mg/kg zanubrutinib vs wild-type treated with DMSO over 10 minutes. Scale bar represents 20 μm. (F) Thrombus volume over time was calculated from thrombus area × thrombus height for all mice treated with ibrutinib, zanubrutinib, or DMSO. (G) Tail bleeding times for wild-type or ibrutinib- or zanubrutinib-treated mice. (H) Volume of blood lost (in microliters) in wild-type or ibrutinib- or zanubrutinib-treated mice. (I) Percentage of unstable hemostasis determined by rebleeding occurring 1 minute after initial clot formation for wild-type or ibrutinib- or zanubrutinib-treated mice. Results are cumulative data from 4 independent experiments and are expressed as the mean ± SEM. **P ≤ .01, ***P ≤ .001, ****P ≤ .0001.
The effect of ibrutinib and zanubrutinib on in vitro, ex vivo, and in vivo thrombus formation and tail bleeding time. (A-B) Fluorescently labeled whole blood of C57BL/6 mice was untreated or treated with 0.5 μM of ibrutinib or 0.5 µM of zanubrutinib and was perfused over 500 μg/mL type I collagen under arterial flow conditions at a shear rate of 1800 seconds−1. Z-stack images were captured over a 6-minute duration with a digital AxioCam MRm camera (Zeiss) and analyzed with Zeiss Axiovision Rel 4.6 software. 3D deconvolved reconstructions of thrombi formed were analyzed for surface coverage of platelet aggregates (in square micrometers), thrombus height (in micrometers), and thrombus volume (in cubic micrometers). (A) Representative images of thrombi formation over type I collagen under arterial flow conditions for wild-type mouse whole blood treated with 0.5 μM of ibrutinib or 0.5 µM of zanubrutinib vs DMSO control over 6 minutes. Scale bar represents 20 μm. (B) Thrombus volume over time was calculated from thrombus area × thrombus height. Results are cumulative data from 4 independent experiments and presented as the mean ± SEM, by the unpaired Student t test. ***P ≤ .005, ****P ≤ .001. (C-D) Fluorescently labeled whole blood of C57BL/6 mice untreated or treated with 10 mg/kg ibrutinib or 10 mg/kg zanubrutinib was perfused over 500 μg/mL type I collagen under arterial flow conditions at a shear rate of 1800 seconds−1. Z-stack images were captured over a 6-minute duration with a digital AxioCam MRm camera (Zeiss) and analyzed with Zeiss Axiovision Rel 4.6 software. 3D deconvolved reconstructions of thrombi formed were analyzed for surface coverage of platelet aggregates, thrombus height, and thrombus volume. Scale bar represents 20 μm. (E) Representative images of thrombus formation over type I collagen under arterial flow conditions for wild-type mice treated with 10 mg/kg ibrutinib or 10 mg/kg zanubrutinib vs wild-type treated with DMSO over 10 minutes. Scale bar represents 20 μm. (F) Thrombus volume over time was calculated from thrombus area × thrombus height for all mice treated with ibrutinib, zanubrutinib, or DMSO. (G) Tail bleeding times for wild-type or ibrutinib- or zanubrutinib-treated mice. (H) Volume of blood lost (in microliters) in wild-type or ibrutinib- or zanubrutinib-treated mice. (I) Percentage of unstable hemostasis determined by rebleeding occurring 1 minute after initial clot formation for wild-type or ibrutinib- or zanubrutinib-treated mice. Results are cumulative data from 4 independent experiments and are expressed as the mean ± SEM. **P ≤ .01, ***P ≤ .001, ****P ≤ .0001.
In ex vivo mouse models, various Btk inhibitors (ibrutinib 10 mg/kg or zanubrutinib 10 mg/kg) were administered orally. The dosing of ibrutinib and zanubrutinib of 10 mg/kg in in vivo mouse experiments is bioequivalent to that in Btk-treated CLL in patients.3 Whole blood was taken 2 hours later (peak drug concentration), rhodamine-labeled, then perfused onto immobilized type I fibrillar collagen at an arterial shear rate of 1800 seconds−1. Thrombus images were recorded in real time over 6 minutes of blood perfusion. Thrombus volume was significantly inhibited by ibrutinib treatment (10 mg/kg) compared with equivalent thrombus growth for zanubrutinib (10 mg/kg) and vehicle-treated control over time (Figure 5C-D). In addition, increased tail bleeding times, associated with an enhanced volume of blood loss and unstable hemostasis, were found in ibrutinib-treated mice but not in wild-type or zanubrutinib-treated mice (Figure 5G-I).
Using in vivo mouse models, various Btk inhibitors (ibrutinib 10 mg/kg or zanubrutinib 10 mg/kg) were administered orally. After 2 hours of incubation, mice were fully anesthetized before cannulation of the jugular vein and exteriorization of the mesenteric arterioles (80-100 µm) was performed. FeCl3-induced vascular injury was undertaken to induce the formation of in vivo blood clots, which was monitored in real time by intravital microscopy. Ibrutinib-treated C57BL/6 mice had smaller thrombus growth over time compared with zanubrutinib- and vehicle control-treated C57BL/6 mice (Figure 5E-F).
Ibrutinib induces shedding of soluble GPIbα and soluble integrin αIIbβ3 from mouse platelets in vivo
To test whether ibrutinib can induce shedding of GPIbα and integrin αIIbβ3 in vivo, we dosed C57BL/6 mice with vehicle, ibrutinib (10 mg/kg), or zanubrutinib (10 mg/kg) by oral gavage for 2 hours, and platelets derived from whole blood were then labeled with anti-mouse CD42b (GPIbα) or anti-mouse CD41 (integrin αIIbβ3) antibodies and analyzed by flow cytometry. Ibrutinib induced loss of GPIbα and integrin αIIbβ3 from murine platelet surfaces, whereas zanubrutinib was comparable to the vehicle control (Figure 6A-B). In addition, plasma levels of shed GPIbα and integrin αIIbβ3 increased 2- and 1.5-fold, respectively, compared with vehicle- or zanubrutinib-treated mice (Figure 6C-D). This result demonstrated that ibrutinib induces shedding of GPIbα and integrin αIIbβ3 in vivo.

Ibrutinib induces shedding of platelet surface GPIbα and integrin αIIbβ3with production of soluble GPIbα and soluble integrin αIIbfrom mouse platelets in vivo. (A-B) Whole blood was collected from C57BL/6 mice treated with 10 mg/kg ibrutinib or 10 mg/kg zanubrutinib or vehicle control after 2 hours of ingestion by oral gavage. Washed murine platelets (100 × 109/L) were then labeled with anti-mouse CD42b (GPIbα) or anti-mouse CD41a (integrin αIIbβ3) PE-conjugated antibodies. Flow cytometric analysis was used to determine the respective platelet glycoprotein expression. Statistical analysis was performed with the Student t test. Results are represented as mean fluorescence intensity (MFI) ± SEM from 3 independent experiments. ****P ≤ .0001. (C-D) Plasma obtained from C57BL/6 mice treated with 10 mg/kg ibrutinib, 10 mg/kg zanubrutinib, or vehicle control was collected after 2 hours of ingestion by oral gavage. Levels of plasma soluble GPIbα (A) and soluble integrin αIIb (B) were determined for vehicle control vs Btk inhibitor-treated C57BL/6 mice using ELISA-based assays. Results represent 3 independent experiments and are plotted as the mean ± SEM, by unpaired Student t test. **P ≤ .01, ***P ≤ .005.
Ibrutinib induces shedding of platelet surface GPIbα and integrin αIIbβ3with production of soluble GPIbα and soluble integrin αIIbfrom mouse platelets in vivo. (A-B) Whole blood was collected from C57BL/6 mice treated with 10 mg/kg ibrutinib or 10 mg/kg zanubrutinib or vehicle control after 2 hours of ingestion by oral gavage. Washed murine platelets (100 × 109/L) were then labeled with anti-mouse CD42b (GPIbα) or anti-mouse CD41a (integrin αIIbβ3) PE-conjugated antibodies. Flow cytometric analysis was used to determine the respective platelet glycoprotein expression. Statistical analysis was performed with the Student t test. Results are represented as mean fluorescence intensity (MFI) ± SEM from 3 independent experiments. ****P ≤ .0001. (C-D) Plasma obtained from C57BL/6 mice treated with 10 mg/kg ibrutinib, 10 mg/kg zanubrutinib, or vehicle control was collected after 2 hours of ingestion by oral gavage. Levels of plasma soluble GPIbα (A) and soluble integrin αIIb (B) were determined for vehicle control vs Btk inhibitor-treated C57BL/6 mice using ELISA-based assays. Results represent 3 independent experiments and are plotted as the mean ± SEM, by unpaired Student t test. **P ≤ .01, ***P ≤ .005.
A proportion of B-CLL patients treated with ibrutinib, but not with zanubrutinib, have reduced GPIbα and integrin αIIbβ3 platelet surface expression and ex vivo thrombus growth
As ibrutinib is associated with shedding of platelet glycoproteins in vivo, we next investigated whether patients with B-CLL treated with ibrutinib had reduced surface expression of GPIbα and integrin αIIbβ3 and ex vivo thrombus formation on type I collagen compared with zanubrutinib-treated patients or healthy controls. Table 1 summarizes information of the Btk inhibitor–treated patients. GPIb-IX-V complex expression on platelets derived from ibrutinib-treated patients with CLL had statistically significant lower levels compared with healthy controls or untreated or zanubrutinib-treated patients (Figure 7A-C). In addition, integrin αIIbβ3 expression on platelets derived from ibrutinib-treated patients with CLL had statistically significant lower levels compared with healthy controls, untreated patients, or zanubrutinib-treated patients. In contrast, platelets derived from ibrutinib-treated patients had similar levels of GPVI expression compared with those from healthy controls, untreated CLL patients, or zanubrutinib-treated patients. There was an exception of 1 zanubrutinib-treated patient with lower GPVI expression. All assessments were performed with normalized platelets from Btk inhibitor–treated patients with B-CLL vs healthy controls. Ibrutinib-treated B-CLL platelets formed smaller thrombi over time compared with zanubrutinib-treated, untreated B-CLL, and healthy normal control platelets (Figure 7D-E). Thrombus volume was reduced in ibrutinib-treated B-CLL platelets at different time points compared with zanubrutinib-treated, untreated, and healthy normal control B-CLL platelets, which showed similar thrombus volume over time. Taken together, these data indicate that ibrutinib- but not zanubrutinib-treated B-CLL patients had reduced thrombus growth on type I collagen, demonstrating an effect of ibrutinib on ex vivo platelet function.
Patient information
| Patient number | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
| Age | 76 | 55 | 70 | 85 | 76 | 76 | 64 | 79 | 54 | 73 | 50 |
| Sex | M | M | M | F | F | M | M | M | M | M | M |
| Diagnosis | Relapsed CLL | Relapsed CLL, 17p | Relapsed CLL, 13q | Relapsed 17p CLL | Upfront SLL | Relapsed CLL 11q | Relapsed WM | Relapsed CLL | CLL | CLL | CLL |
| Hb, g/L | 122 | 140 | 131 | 116 | 120 | 148 | 177 | 140 | 143 | 159 | 140 |
| Platelets, × 109/L | 88 | 196 | 99 | 179 | 213 | 73 | 217 | 146 | 179 | 113 | 171 |
| Concurrent medication | Aspirin | None | Plavix 75 mg qd | None | Mobic 15 mg prn | None | Aspirin | None | None | None | None |
| Duration of Btk therapy | 19 mo, IB 420 mg qd | 55 mo, IB 420 mg qd | 29 mo, IB 420 mg qd | 55 mo, IB 420 mg qd | 19.5 mo, BGB 160 mg bid | 31 mo, IB 420 mg qd | 36 mo, BGB 160 mg bid | 24 mo, BGB 160 mg bid | 67 mo, IB | 42 mo, BGB | 33 mo, BGB |
| Active bleeding at time of test | None | None | None | None | None | None | None | None | None | None | None |
| Bleeding with Btk treatment | Grade 1 easy bruising | None | Grade 1 bruising | Grade 2 bleeding | Grade 1 bruising | None | None | None | Grade 1 bruising | Grade 1 bruising | Grade 1 bruising |
| Disease response | PR | PR | CR | CR | PR | CR | PR | PR | CR | CR | PR |
| Patient number | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
| Age | 76 | 55 | 70 | 85 | 76 | 76 | 64 | 79 | 54 | 73 | 50 |
| Sex | M | M | M | F | F | M | M | M | M | M | M |
| Diagnosis | Relapsed CLL | Relapsed CLL, 17p | Relapsed CLL, 13q | Relapsed 17p CLL | Upfront SLL | Relapsed CLL 11q | Relapsed WM | Relapsed CLL | CLL | CLL | CLL |
| Hb, g/L | 122 | 140 | 131 | 116 | 120 | 148 | 177 | 140 | 143 | 159 | 140 |
| Platelets, × 109/L | 88 | 196 | 99 | 179 | 213 | 73 | 217 | 146 | 179 | 113 | 171 |
| Concurrent medication | Aspirin | None | Plavix 75 mg qd | None | Mobic 15 mg prn | None | Aspirin | None | None | None | None |
| Duration of Btk therapy | 19 mo, IB 420 mg qd | 55 mo, IB 420 mg qd | 29 mo, IB 420 mg qd | 55 mo, IB 420 mg qd | 19.5 mo, BGB 160 mg bid | 31 mo, IB 420 mg qd | 36 mo, BGB 160 mg bid | 24 mo, BGB 160 mg bid | 67 mo, IB | 42 mo, BGB | 33 mo, BGB |
| Active bleeding at time of test | None | None | None | None | None | None | None | None | None | None | None |
| Bleeding with Btk treatment | Grade 1 easy bruising | None | Grade 1 bruising | Grade 2 bleeding | Grade 1 bruising | None | None | None | Grade 1 bruising | Grade 1 bruising | Grade 1 bruising |
| Disease response | PR | PR | CR | CR | PR | CR | PR | PR | CR | CR | PR |
bid, twice daily; BGB, zanubrutinib; CR, complete remission; F, female; IB, ibrutinib; M, male; PR, partial remission; prn, taken on an as-needed basis; qd, daily; SLL, small lymphocytic lymphoma; WM, Waldenström macroglobulinemia.
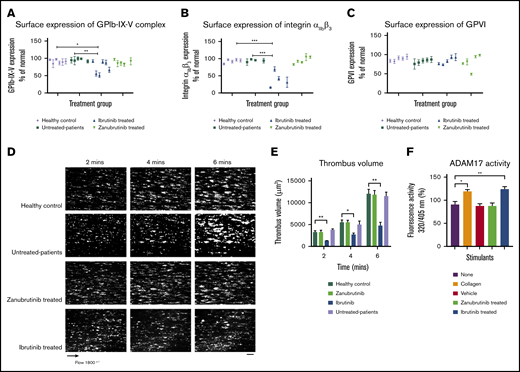
Ibrutinib but not zanubrutinib treatment leads to loss of GPIb-IX-V complex and integrin αIIbβ3 from the platelet surface and reduced ex vivo thrombus formation on type I collagen during arterial flow in B-CLL. Washed B-CLL platelets (100 × 109/L) on either untreated or ibrutinib or zanubrutinib or normal healthy donor platelets were labeled with anti-human CD42a, anti-human CD41a, or anti-human GPVI PE-conjugated antibodies. Flow cytometric analysis was used to determine GPIb-IX (A), integrin αIIbβ3 (B), and GPVI (C) expression. Results are expressed as the mean ± SEM from a single collection tested in replicate and represent 3 independent experiments, by Student t test. *P ≤ .05, **P ≤ .01, ***P ≤ .005. (D) Representative images of thrombus formation over type I collagen in arterial flow conditions for patients with B-CLL whole blood, either untreated or treated with ibrutinib or zanubrutinib vs healthy control over 6 minutes. Scale bar represents 20 μm. (E) Thrombus volume (in cubic micrometers) over time was calculated by thrombus area (in square micrometers) × thrombus height (in micrometers) for B-CLL patients, untreated or treated with ibrutinib, zanubrutinib, or healthy control. Results are expressed as the mean ± SEM from a single collection tested in replicate. (F) Washed human platelets (200 µL of 100 × 109/L) were pretreated with 20 µM fluorogenic TACE substrate at 37°C for 20 minutes. Platelets were then incubated with either vehicle control, 20 µg/mL type I collagen, 0.5 µM ibrutinib, or 0.5 µM zanubrutinib at 37°C for 40 minutes. The cleavage of the TACE substrate was monitored at 320/405 nm using a Clariostar microplate reader. Results represent 3 independent experiments and are plotted as the mean ± SEM, by unpaired Student t test. **P ≤ .01, ***P ≤ .005.
Ibrutinib but not zanubrutinib treatment leads to loss of GPIb-IX-V complex and integrin αIIbβ3 from the platelet surface and reduced ex vivo thrombus formation on type I collagen during arterial flow in B-CLL. Washed B-CLL platelets (100 × 109/L) on either untreated or ibrutinib or zanubrutinib or normal healthy donor platelets were labeled with anti-human CD42a, anti-human CD41a, or anti-human GPVI PE-conjugated antibodies. Flow cytometric analysis was used to determine GPIb-IX (A), integrin αIIbβ3 (B), and GPVI (C) expression. Results are expressed as the mean ± SEM from a single collection tested in replicate and represent 3 independent experiments, by Student t test. *P ≤ .05, **P ≤ .01, ***P ≤ .005. (D) Representative images of thrombus formation over type I collagen in arterial flow conditions for patients with B-CLL whole blood, either untreated or treated with ibrutinib or zanubrutinib vs healthy control over 6 minutes. Scale bar represents 20 μm. (E) Thrombus volume (in cubic micrometers) over time was calculated by thrombus area (in square micrometers) × thrombus height (in micrometers) for B-CLL patients, untreated or treated with ibrutinib, zanubrutinib, or healthy control. Results are expressed as the mean ± SEM from a single collection tested in replicate. (F) Washed human platelets (200 µL of 100 × 109/L) were pretreated with 20 µM fluorogenic TACE substrate at 37°C for 20 minutes. Platelets were then incubated with either vehicle control, 20 µg/mL type I collagen, 0.5 µM ibrutinib, or 0.5 µM zanubrutinib at 37°C for 40 minutes. The cleavage of the TACE substrate was monitored at 320/405 nm using a Clariostar microplate reader. Results represent 3 independent experiments and are plotted as the mean ± SEM, by unpaired Student t test. **P ≤ .01, ***P ≤ .005.
Ibrutinib, but not zanubrutinib, increased ADAM17 TACE activity in vitro
To investigate why ibrutinib-induced shedding of GPIbα and GPIX occurs in an ADAM17-dependent manner, we used a TACE substrate to examine the effect of ibrutinib compared with zanubrutinib and vehicle control on ADAM17 TACE activity in vitro. Ibrutinib treatment of washed platelets resulted in increased TACE activity in vitro compared with the effect of zanubrutinib or vehicle control treatment (Figure 7F).
Discussion
Before our study, it was thought that ibrutinib inhibits a combination of collagen GPVI signaling and outside-in integrin αIIbβ3 signaling to reduce the platelet function and thrombus formation that contribute to bleeding risk.16,20 Here, we report that ibrutinib induces the proteolytic cleavage of human and murine platelet surface receptors, including GPIbα, GPIX, and integrin αIIbβ3 in vitro and in vivo. In the case of GPIbα, ibrutinib increases activation of the metalloproteinase ADAM17 and mediates proteolytic cleavage of GPIbα and GPIX, but not of GPV or GPIbβ. In contrast, integrin αIIbβ3 is shed by an unknown protease. These processes are relevant in vivo, because mice given ibrutinib by oral gavage had reduced ex vivo and in vivo thrombus formation, prolonged tail bleeding times, and increased levels of shed GPIbα and platelet-specific integrin αIIb in their plasma.
In vitro thrombus formation experiments demonstrated that ibrutinib reduced platelet adhesion and thrombus formation on immobilized type I collagen, VWF, and fibrinogen. These adhesive interactions are involved in direct collagen and indirect collagen receptors, GPIb-IX-V complex, and integrin αIIbβ3. This was not the case for the more selective Btk inhibitor zanubrutinib, and it raised the possibility that ibrutinib modulates platelet adhesion and thrombus formation by a different mechanism, apart from modulating GPVI signaling or outside-in integrin αIIbβ3 signaling in platelets. We therefore investigated the surface expression of platelet glycoproteins in vitro and in vivo and found that ibrutinib treatment induced loss of GPIbα, GPIX, and integrin αIIbβ3, but not of GPVI, GPV, or GPIbβ. This raised the possibility that either proteolytic shedding or internalization occurs in vitro or in vivo. To address these possibilities, we examined a range of inhibitors, including the broad-spectrum metalloproteinase inhibitor GM6001, the ADAM17 inhibitor TAPI-2, the ADAM10 inhibitor GI254023, the calpain inhibitor calpeptin, the dynamin inhibitor Dynasore hydrate, and the integrin αIIbβ3 antagonist eptifibatide, to see whether they would block the loss of either GPIbα or integrin αIIbβ3 from the platelet surface. Our results showed that the ADAM17 inhibitor TAPI-2 blocked the ibrutinib-induced shedding of GPIbα and GPIX from the platelet surface. This was not the case with the ADAM10 inhibitor GI254023, the calpain inhibitor calpeptin, the dynamin inhibitor Dynasore hydrate, and the integrin αIIbβ3 antagonist eptifibatide. In contrast, the ADAM17 inhibitor TAPI-2, the ADAM10 inhibitor GI254023, the calpain inhibitor calpeptin, or the integrin αIIbβ3 antagonist eptifibatide did not block the ibrutinib-induced shedding of integrin αIIbβ3 from the platelet surface.
The mechanism by which ibrutinib exerts its action is mainly attributable to an off-target effect where metalloproteinase ADAM17 activity is increased by the protease’s being quickly switched on when platelets are activated to induce proteolytic shedding of GPIbα from the platelet surface. In addition, ibrutinib appears to affect an unknown sheddase to induce proteolytic shedding of integrin αIIbβ3 from the platelet surface. This hypothesis is based on the fact that zanubrutinib targets Btk as potently as ibrutinib but appears to be more selective in its Btk inhibition without the observed effects on platelet function and receptor shedding.
In platelets, the predominant sheddases are ADAM10 and ADAM17, with ADAM17 showing significant effects on a range of platelet receptors including GPIX and, exclusively, GPIbα and GPV.21,22 ADAM10 activity plays an important role in downregulating GPVI levels on platelets.21 Under physiological conditions, GPIbα is constitutively shed as a glycocalicin fragment, with 2 µg/mL present in human plasma, and is thought to be important as a measure of platelet turnover.21 High-dose aspirin treatment has been shown to induce ADAM17-mediated GPIbα shedding in vitro and in vivo.28 The significance of this process is not well understood, but very likely indicates downregulation of the inflammatory response and platelet responsiveness.
In this study, we demonstrated that ibrutinib, a Btk inhibitor, appears to have off-target effects that induce ADAM17-mediated shedding of GPIbα and GPIX in vitro and of GPIbα in vivo, as a soluble glycocalicin fragment detected in mouse plasma. This was not the case with the more selective Btk inhibitor zanubrutinib. The shedding process was not dependent on ADAM10 or calpain cleavage, nor was it regulated by outside-in integrin αIIbβ3 signaling. The mechanism underlying this ibrutinib effect is not completely clear, but it was shown that ibrutinib treatment increased ADAM17 TACE activity compared with zanubrutinib and vehicle control in vitro. Under physiological conditions, proteolytic cleavage of the prodomain activates the ADAM family members; however, direct activation of ADAM sheddases can occur with thiol-modifying agents or calmodulin inhibitors to induce GPIbα shedding in vitro.29 The precise mechanism of how ibrutinib induces GPIbα and GPIX shedding requires further investigation, but the increased activation of ADAM17 may be related to the off-target effects of ibrutinib on 1 or more kinases, including protein kinase C, p38 kinase, or pololike kinase. Alternatively, ibrutinib is known to have potential off-target effects with 9 other cysteine-containing kinases in the human kinome that have cysteine at a similar position within the ATP binding pocket (Bmx, Tec, Txk, Itk, EGFR, Erb2, Erb4, Jak3, and Blk).
Apart from GPIbα, in this study ibrutinib also induced shedding of integrin αIIbβ3 from the surface of platelets in vitro and in vivo. However, in contrast to GPIbα and GPIX, it was independent of ADAM17-, ADAM10-, or calpain-induced shedding and was not regulated by outside-in integrin αIIbβ3 signaling. Analysis of the platelet sheddome highlighted that integrin αIIb and β3 subunits were detected in the supernatant of activated platelets, indicating that they can be shed from the surface of platelets.22 The mechanism of how this shedding process occurs with integrin αIIbβ3 is unknown. Previous studies have highlighted a role for calpain cleavage of the intracellular cytoplasmic domain of integrin β3, and high shear forces can induce shedding of integrin αIIbβ3.30,31 Although ADAM17 has been shown to have mild effects on integrin αIIbβ3, it was excluded as a possible mechanism in the current study.22 Whereas calpain cleavage may have also contributed to ectodomain shedding, calpeptin treatment before ibrutinib treatment did not block loss of integrin αIIbβ3 from the platelet surface.
The likely explanation for the loss of integrin αIIbβ3 from the surface of platelets after ibrutinib treatment may be attributable to induction of internalization or shedding of the receptor. Blocking internalization with a dynamin inhibitor, Dynasore hydrate before ibrutinib treatment did not reverse the ibrutinib-mediated reduction in integrin αIIbβ3 surface expression. However, in our mouse model, we were able to demonstrate that platelet-specific soluble integrin αIIb was present in increased amounts in plasma after exposure to ibrutinib in vivo compared with zanubrutinib and vehicle control, supporting the concept that shedding is associated with loss of integrin αIIbβ3 from the platelet surface. These experiments highlighted that ibrutinib treatment induces integrin αIIbβ3 shedding from the platelet surface by an unknown sheddase. Further studies are needed to determine the exact sheddase involved in integrin αIIbβ3 ectodomain shedding in vivo.
Apart from in vitro and in vivo experiments, we also examined B-CLL patient samples from untreated or ibrutinib- vs zanubrutinib-treated and normal healthy controls and showed a significant proportion of ibrutinib-treated samples but not zanubrutinib or healthy controls had reduced surface expression of GPIbα, GPIX, and integrin αIIbβ3. In addition, we demonstrated that ibrutinib-treated patients with B-CLL had reduced ex vivo thrombus formation on type I collagen during arterial flow compared with zanubrutinib-treated patients, who were equivalent to untreated patients and normal healthy controls. This finding indicates that the shedding of major platelet receptors affects platelet function in ibrutinib-treated B-CLL patients and can explain why these patients are at risk of major bleeding. In this context, for patients with B-CLL, zanubrutinib may be safer in terms of bleeding risk for treating their hematological malignancy while maintaining a primary hemostatic balance with milder adverse events of petechiae/bruising reported in 38% of cases,32 whereas acalabrutinib is associated with bleeding events in ∼50% of patients with hematological malignancies.33 This is most likely attributable to on-target inhibition of collagen GPVI signaling pathways.
Other workers have studied the effect of ibrutinib on human platelets in modulating collagen- and VWF-dependent functions12 and integrin αIIbβ3 outside-in signaling.20 We also observed that ibrutinib affected collagen- and VWF-dependent functions and outside-in integrin αIIbβ3 signaling readouts. However, our study showed that a quantitative reduction in GPIb-IX and integrin αIIbβ3 associated with shedding mediated by ibrutinib treatment contributed to the qualitative functional abnormalities observed in these previous studies.
In summary, in the current study, ibrutinib, but not zanubrutinib, caused shedding of human and mouse GPIbα, and GPIX in vitro and in vivo from the platelet surface. This process involves increased activation of the metalloproteinase ADAM17 and is exclusively associated with ADAM17. In addition, we have shown that ibrutinib but not zanubrutinib induces shedding of human and mouse integrin αIIbβ3 in vitro and in vivo by an unknown sheddase. These results are important, as they are a like explanation of why patients with ibrutinib-treated B-CLL have a higher risk of bleeding complications. At this stage, zanubrutinib is not yet approved for use in the United States, and acalabrutinib has been approved by the US Food and Drug Administration only for mantle cell lymphoma. Until these more selective Btk inhibitors are approved in more settings, ibrutinib will continue to be the dominant Btk inhibitor in clinical use.
Acknowledgments
This work was supported by a sponsored research agreement from Beigene, Pty Ltd (D.E.J.); funding from CLL Global Research Foundation (C.T.); and a postgraduate scholarship from Jazan University and the Ministry of Higher Education (Riyadh, Saudi Arabia) (G.D.).
Authorship
Contribution: G.D., F.A.K., M.M.A.O., F.A., and A.M.G. performed the experiments; D.M.-y.S., S.M.H., C.P.S.T., and C.T. provided intellectual input and reviewed the paper; and D.E.J. directed the research and wrote the manuscript.
Conflict-of-interest disclosure: Beigene Pty, Ltd, provides funding to C.T. and D.E.J. The remaining authors declare no competing financial interests.
Correspondence: Denise E. Jackson, Thrombosis and Vascular Diseases Laboratory, School of Health and Biomedical Sciences, RMIT University, P.O. Box 71, Bundoora, VIC 3083, Australia; e-mail: denise.jackson@rmit.edu.au.

