

Key Points
In adults undergoing allo-HSCT, vedolizumab IV 300 mg was well tolerated and had a low incidence of overall and lower-intestinal aGVHD.
These phase 1b data support further evaluation of vedolizumab for the prevention of aGVHD in adults undergoing allo-HSCT.

Abstract
Acute graft-versus-host disease (aGVHD) remains a significant complication of allogeneic hematopoietic stem cell transplantation (allo-HSCT). Vedolizumab could help prevent aGVHD by inhibiting the migration of both naive and activated lymphocytes into gut-associated lymphoid tissues and the lamina propria. We carried out a phase 1b, open-label, dose-finding study in adults undergoing allo-HSCT to evaluate the tolerability, safety, and pharmacokinetics of vedolizumab, and its effectiveness in reducing aGVHD. IV vedolizumab was administered on day −1, +13, and +42 with respect to allo-HSCT, starting at 75 mg and with dose escalation guided by tolerability and pharmacokinetics. A total of 24 participants was enrolled, and no dose-limiting toxicities were observed in either the 75-mg cohort (n = 3) or the dose-escalated 300-mg cohort (n = 21). Treatment-emergent adverse events related to vedolizumab occurred in 8 participants. Overall, 4 deaths occurred during the 12 months following allo-HSCT. No participants in the 75-mg cohort developed modified Glucksberg grade II to IV aGVHD by 100 days after allo-HSCT. Four participants (19.0%) in the 300-mg cohort developed grade II to IV aGVHD by 100 days after allo-HSCT, including 3 participants who developed stage 1 aGVHD of the lower-intestinal tract. Vedolizumab IV 300 mg was well tolerated as aGVHD prevention, and the incidence of overall and lower-intestinal aGVHD was low. These findings support further evaluation of vedolizumab in this patient population. This trial was registered at www.clinicaltrials.gov as #NCT02728895.
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is a potentially curative therapy for several hematologic malignancies. Despite current prophylaxis measures, acute graft-versus-host disease (aGVHD) remains a major complication after allo-HSCT and is associated with significant morbidity and mortality.1-3 Patients undergoing allo-HSCT have an incidence of grade II to IV aGVHD of ∼40% to 70%.4-6 The risk of developing aGVHD following allo-HSCT is variable, depending on the degree of histocompatibility between donor and recipient, GVHD prophylaxis regimen employed, recipient and donor clinical factors, as well as intensity and type of conditioning regimen used.7,8
Acute GVHD affects the skin, gut, and liver, with aGVHD of the lower-intestinal tract resulting in most of the morbidity and mortality.2,3,9 Allogeneic donor T cells stimulated by the presence of recipient alloantigens represent a key feature in the pathophysiology of aGVHD. In addition, inflammation of and damage to the intestinal tract is a major driver for the amplification of systemic aGVHD.2,3,9,10 α4β7 integrin, expressed on gut-homing T lymphocytes, is a pivotal mediator of gut immunity and inflammation. It has a central role in mediating the migration of both naive and activated lymphocytes into gut-associated lymphoid tissues and the lamina propria, via its binding to mucosal addressin cell adhesion molecule 1 (MAdCAM-1).11-14 Studies in mouse models suggest that absence of T-cell trafficking to gut-associated lymphoid tissue via inhibition of the α4β7 integrin/MAdCAM-1 interaction may prevent aGVHD.15-17 Similarly, α4β7 integrin expression has been shown to be significantly increased on naive and memory T cells in patients who underwent allo-HSCT and developed intestinal aGVHD, compared with patients who either developed aGVHD of the skin or had no aGVHD.18
Vedolizumab is a humanized monoclonal antibody that specifically targets α4β7 integrin and inhibits its adhesion to MAdCAM-1, thereby demonstrating gut-selective immunomodulatory activity. As such, vedolizumab is approved for the treatment of moderate to severe active ulcerative colitis and Crohn disease in adults. Given its specific mechanism of action and established safety profile in patients with inflammatory bowel disease, we hypothesized that vedolizumab would have acceptable tolerability in patients undergoing allo-HSCT and potentially could help prevent lower-intestinal aGVHD. We investigated the clinical activity of vedolizumab when added to standard GVHD prophylaxis in patients undergoing allo-HSCT and evaluated its tolerability, safety, pharmacokinetics, and preliminary efficacy in reducing the incidence of aGVHD.
Patients and methods
Study design
This was a phase 1b, open-label, dose-finding study (clinical trial registration: #NCT02728895) in which vedolizumab was added to standard GVHD prophylaxis (tacrolimus plus short-term methotrexate [MTX]) in adult participants undergoing allo-HSCT at 5 US sites.
Vedolizumab dosing followed a rule-based dose-finding study design with pharmacokinetic (PK) guidance and comprised 2 parts: an initial dose-finding study to establish a dose with an acceptable PK profile followed by an expansion phase to further assess the tolerability and clinical activity of vedolizumab (more details in “Study procedures”).
This study was approved by each site’s institutional review board, and all participants provided informed consent according to the principles of the Declaration of Helsinki and the International Council for Harmonization Guidelines for Good Clinical Practice.19,20
The primary objective was to describe the initial tolerability and safety of vedolizumab, and to identify a recommended dose when added to standard GVHD prophylaxis in participants undergoing allo-HSCT. Secondary objectives included characterization of the PK profile of vedolizumab in participants on days −1, +13, and +42 days after allo-HSCT, and determination of the cumulative incidence and the severity of aGVHD in participants by 100 days after allo-HSCT.
Study participants
Inclusion and exclusion criteria were specific for the dose-finding and expansion phases. For the dose-finding phase, the main inclusion criteria were ages 18 to 60 years; candidate for HLA-matched unrelated donor or 1-locus (antigen or allele) HLA-mismatched unrelated donor allo-HSCT, using peripheral blood or bone marrow hematopoietic stem cell sources, an underlying diagnosis of acute lymphoblastic leukemia (ALL) or acute myeloid leukemia (AML) both in remission defined as <5% blast cells, hematopoietic recovery (complete remission with incomplete platelet recovery allowed); no evidence of extramedullary disease; and use of myeloablative conditioning.21 For the expansion phase, the main inclusion criteria were ages 18 to 60 years for those undergoing myeloablative conditioning, or ages 18 to 75 years for those undergoing reduced-intensity conditioning; a candidate for HLA-matched or 1-locus (antigen or allele) HLA-mismatched related or unrelated donor allo-HSCT, using peripheral blood or bone marrow hematopoietic stem cell sources; and an underlying diagnosis of hematologic malignancy or myeloproliferative neoplasm with <5% bone marrow blasts at time of allo-HSCT. For both phases, exclusion criteria included history of any major neurological disorders and active systemic infection. Full details of inclusion and exclusion criteria are given in supplemental Table 1.
Study procedures
All participants received either a myeloablative or a reduced intensity conditioning regimen, followed by tacrolimus (recommended goal serum trough concentration of 5 to 10 ng/dL) and MTX (recommended 10 mg/m2 IV on days +1, +3, +6, and +11 after allo-HSCT). Participants received open-label vedolizumab as an IV infusion on day −1 before allo-HSCT and then on day +13 and day +42 after allo-HSCT.
Dose-finding phase
Dose escalation was guided by the monitoring of dose-limiting toxicities (DLTs), and toxicity was evaluated according to the Common Terminology Criteria for Adverse Events, Version 4.03.22 A DLT was defined as any grade III or higher toxicity assessed by the investigator as related to the study drug. Failure to engraft by day +28 was considered a DLT. Hypersensitivity reactions and other infusion-related reactions were not considered DLTs. Grade IV laboratory abnormalities that were not considered clinically significant by the investigator were not considered DLTs.
Dose escalation started with a low-dose cohort where participants received 75 mg vedolizumab IV on day −1, +13, and +42. Dosing began with the first participant, who received 75 mg vedolizumab IV and was monitored for DLTs from the start of the first IV infusion of vedolizumab on day −1 to day +28 (DLT observation period) after allo-HSCT. If this participant tolerated vedolizumab IV at 75 mg and engraftment occurred (recovery of absolute neutrophil count, absolute neutrophil count >0.5 ×109/L for 3 days consecutively or >2 ×109/L for 1 day), then 2 more participants were enrolled and observed for DLTs. If none of the 3 participants of the low-dose cohort experienced DLTs, the next cohort would receive vedolizumab IV at 300 mg. Based on previous analyses of the 300-mg dose of vedolizumab IV in participants with ulcerative colitis and Crohn disease,23 the serum concentration of vedolizumab was expected to maintain an α4β7 integrin receptor saturation of >90% over the dosing interval in >90% of participants. If the first participant in the 300-mg dose cohort tolerated vedolizumab at this dose and engraftment occurred, then 2 more participants were enrolled in the cohort. If the first 3 participants at 300 mg tolerated the treatment without experiencing DLTs, then the decision of whether to increase the vedolizumab dose above 300 mg in the next cohort was guided by the PK results. If any participant in each cohort experienced a DLT, then 3 additional participants would be enrolled at the same dose level and monitored for DLTs from day −1 until day +28.
Expansion phase
After a tolerated dose with acceptable PK results was identified, the cohort at that dose level was expanded to include ∼18 additional participants.
Assessment and follow-up periods
Assessment of DLTs was carried out over a 30-day period from day −1 to day +28, and PK measurements were collected throughout the first 100 days following allo-HSCT. Treatment-emergent adverse events (TEAEs) were monitored during the treatment-emergent period: from day −1 to 18 weeks following the last dose of vedolizumab. Participants were followed for overall survival, safety, and development of aGVHD for 12 months after allo-HSCT or until the participant’s death or withdrawal, whichever occurred first.
Disease assessment
Acute GVHD was assessed using the modified Glucksberg criteria.24
PK and immunogenicity measurements
Serial blood samples for PK evaluation and determination of serum concentrations of vedolizumab were obtained at multiple time points from day −1 through day +100 (supplemental Table 2). The presence of anti–vedolizumab antibodies (AVAs) in serum was also evaluated.
Outcomes
The primary endpoints included frequency of DLTs during the DLT observation period, and the number and percentage of participants who experienced TEAEs and serious AEs. Secondary endpoints comprised time to neutrophil engraftment; percentage of participants who developed overall grade II to IV (according to modified Glucksberg criteria)24,25 aGVHD by day +100 after allo-HSCT; the frequency of events, by maximum severity of aGVHD; and mean serum concentrations of vedolizumab before dosing (Ctrough) on days +13 and +42 after allo-HSCT.
Exploratory endpoints included percentage of participants who developed skin, intestinal tract, or liver aGVHD; overall survival at 12 months; and nonrelapse mortality (NRM, the survival in the absence of primary malignancy relapse after allo-HSCT) at 12 months.
Statistical analysis
The safety population consisted of all participants who received any amount of vedolizumab, and the PK population consisted of participants from the safety population with at least 1 sample collected for PK analysis. For the primary endpoint, an evaluable participant was one who received vedolizumab and was assessed for engraftment on or before day +28. Demographic and baseline characteristics were summarized by dose cohort for the safety population. For PK analyses, measured serum concentrations of vedolizumab and other PK parameters (area under the concentration–time curve [AUC]d1-13, AUCd13-42, AUCd42-100, and maximum observed concentration [Cmax]) were summarized using descriptive statistics by dose cohort and treatment time, separately, and percentages of participants who were AVA-positive and AVA-negative were computed. Safety was evaluated by the incidence of adverse events (AEs), and severity and type of AEs, summarized by dose cohort. AEs were tabulated according to the Medical Dictionary for Regulatory Activities (MedDRA) by system organ class. Efficacy endpoints were evaluated by incidence of aGVHD by 100 days after allo-HSCT, summarized by dose cohort, using the safety population. Time-to-event (survival) endpoints were summarized for each dosing cohort. NRM was estimated using cumulative incidence function in the presence of competing risk. Kaplan-Meier estimates of event rates (n, %) at 6 and 12 months were computed along with Kaplan-Meier survival curves. No imputation of values for missing data was performed.
Results
Characteristics of study participants
The final study sample comprised a total of 24 participants with a median (range) age of 55 (18 to 72) years with clinical characteristics listed in Table 1. Three participants enrolled in the initial 75-mg dose cohort and experienced no DLTs. Three participants were then enrolled in the 300-mg dose cohort and experienced no DLTs, and an additional 18 participants were enrolled (as planned) in the 300-mg dose cohort (n = 21, 300-mg dose cohort). All participants (N = 24) completed the study up to day +28 (supplemental Figure 1). Overall, 13 participants (54.2%) received a myeloablative conditioning regimen, and 20 participants (83.3%) had an unrelated donor transplant (Table 1). Nine participants (37.5%) received hematopoietic stem cells from bone marrow, whereas 15 participants (62.5%) received peripheral blood grafts. All participants in the 75-mg dose cohort underwent transplantation using bone marrow as the cell source.
Baseline characteristics of study sample, by dose cohort
| Characteristic | Vedolizumab 75 mg (n = 3) | Vedolizumab 300 mg (n = 21) | Total (N = 24) |
|---|---|---|---|
| Median age (range), y | 22.0 (18-50) | 58.0 (19-72) | 55.0 (18-72) |
| Male | 1 (33.3) | 16 (76.2) | 17 (70.8) |
| Mean weight (SD), kg | 92.93 (25.66) | 89.96 (25.84) | 90.33 (25.27) |
| Baseline ECOG performance status | |||
| 0 | 2 (66.7) | 8 (38.1) | 10 (41.7) |
| 1 | 1 (33.3) | 12 (57.1) | 13 (54.2) |
| 2 | 0 (0) | 1 (4.8) | 1 (4.2) |
| Disease type | |||
| Myeloproliferative neoplasm | 0 (0) | 3 (14.3) | 3 (12.5) |
| Myelodysplastic/myeloproliferative neoplasm | 0 (0) | 3 (14.3) | 3 (12.5) |
| Myelodysplastic syndrome | 0 (0) | 2 (9.5) | 2 (8.3) |
| AML or related precursor neoplasm | 3 (100) | 6 (28.6) | 9 (37.5) |
| Precursor lymphoid neoplasm | 0 (0) | 5 (23.8) | 5 (20.8) |
| Precursor T-ALL/LBL | 0 (0) | 3 (14.3) | 3 (12.5) |
| Precursor B-ALL/LBL | 0 (0) | 2 (9.5) | 2 (8.3) |
| Other | 0 (0) | 2 (9.5) | 2 (8.3) |
| Conditioning regimen | |||
| Myeloablative, busulfan + fludarabine | 2 (66.7) | 5 (23.8) | 7 (29.2) |
| Myeloablative, cyclophosphamide + TBI | 1 (33.3) | 5 (23.8) | 6 (25.0) |
| Reduced-intensity, busulfan + fludarabine | 0 (0) | 6 (28.6) | 6 (25.0) |
| Reduced-intensity, fludarabine + melphalan | 0 (0) | 5 (23.8) | 5 (20.8) |
| Source of stem cells | |||
| Bone marrow | 3 (100) | 6 (28.6) | 9 (37.5) |
| Peripheral blood | 0 (0) | 15 (71.4) | 15 (62.5) |
| Donor-recipient sex match | |||
| Female-male/male-female | 2 (66.7) | 9 (42.9) | 11 (45.8) |
| Other | 1 (33.3) | 12 (57.1) | 13 (54.2) |
| Donor relationship to study participant | |||
| Related | 0 (0) | 4 (19.0) | 4 (16.7) |
| Unrelated | 3 (100) | 17 (81.0) | 20 (83.3) |
| HLA compatibility | |||
| Matched | 3 (100) | 20 (95.2) | 23 (95.8) |
| Mismatched | 0 (0) | 1 (4.8) | 1 (4.2) |
| Donor CMV IgG antibody | |||
| Positive | 0 (0) | 10 (47.6) | 10 (41.7) |
| Negative | 3 (100) | 11 (52.4) | 14 (58.3) |
| HCT comorbidity index | |||
| Yes | 2 (66.7) | 17 (81.0) | 19 (79.2) |
| <4 | 1 (33.3) | 13 (61.9) | 14 (58.3) |
| ≥4 | 1 (33.3) | 4 (19.0) | 5 (20.8) |
| No or unknown | 1 (33.3) | 4 (19.0) | 5 (20.8) |
| Characteristic | Vedolizumab 75 mg (n = 3) | Vedolizumab 300 mg (n = 21) | Total (N = 24) |
|---|---|---|---|
| Median age (range), y | 22.0 (18-50) | 58.0 (19-72) | 55.0 (18-72) |
| Male | 1 (33.3) | 16 (76.2) | 17 (70.8) |
| Mean weight (SD), kg | 92.93 (25.66) | 89.96 (25.84) | 90.33 (25.27) |
| Baseline ECOG performance status | |||
| 0 | 2 (66.7) | 8 (38.1) | 10 (41.7) |
| 1 | 1 (33.3) | 12 (57.1) | 13 (54.2) |
| 2 | 0 (0) | 1 (4.8) | 1 (4.2) |
| Disease type | |||
| Myeloproliferative neoplasm | 0 (0) | 3 (14.3) | 3 (12.5) |
| Myelodysplastic/myeloproliferative neoplasm | 0 (0) | 3 (14.3) | 3 (12.5) |
| Myelodysplastic syndrome | 0 (0) | 2 (9.5) | 2 (8.3) |
| AML or related precursor neoplasm | 3 (100) | 6 (28.6) | 9 (37.5) |
| Precursor lymphoid neoplasm | 0 (0) | 5 (23.8) | 5 (20.8) |
| Precursor T-ALL/LBL | 0 (0) | 3 (14.3) | 3 (12.5) |
| Precursor B-ALL/LBL | 0 (0) | 2 (9.5) | 2 (8.3) |
| Other | 0 (0) | 2 (9.5) | 2 (8.3) |
| Conditioning regimen | |||
| Myeloablative, busulfan + fludarabine | 2 (66.7) | 5 (23.8) | 7 (29.2) |
| Myeloablative, cyclophosphamide + TBI | 1 (33.3) | 5 (23.8) | 6 (25.0) |
| Reduced-intensity, busulfan + fludarabine | 0 (0) | 6 (28.6) | 6 (25.0) |
| Reduced-intensity, fludarabine + melphalan | 0 (0) | 5 (23.8) | 5 (20.8) |
| Source of stem cells | |||
| Bone marrow | 3 (100) | 6 (28.6) | 9 (37.5) |
| Peripheral blood | 0 (0) | 15 (71.4) | 15 (62.5) |
| Donor-recipient sex match | |||
| Female-male/male-female | 2 (66.7) | 9 (42.9) | 11 (45.8) |
| Other | 1 (33.3) | 12 (57.1) | 13 (54.2) |
| Donor relationship to study participant | |||
| Related | 0 (0) | 4 (19.0) | 4 (16.7) |
| Unrelated | 3 (100) | 17 (81.0) | 20 (83.3) |
| HLA compatibility | |||
| Matched | 3 (100) | 20 (95.2) | 23 (95.8) |
| Mismatched | 0 (0) | 1 (4.8) | 1 (4.2) |
| Donor CMV IgG antibody | |||
| Positive | 0 (0) | 10 (47.6) | 10 (41.7) |
| Negative | 3 (100) | 11 (52.4) | 14 (58.3) |
| HCT comorbidity index | |||
| Yes | 2 (66.7) | 17 (81.0) | 19 (79.2) |
| <4 | 1 (33.3) | 13 (61.9) | 14 (58.3) |
| ≥4 | 1 (33.3) | 4 (19.0) | 5 (20.8) |
| No or unknown | 1 (33.3) | 4 (19.0) | 5 (20.8) |
Data are presented as n (%) unless otherwise stated.
ECOG, Eastern Cooperative Oncology Group; HCT, hematopoietic cell transplantation; IgG, immunoglobulin G; LBL, lymphoblastic lymphoma; SD, standard deviation; TBI, total body irradiation.
DLTs and engraftment
No DLTs were observed for participants in either dose cohort (75 mg and 300 mg). Neutrophil engraftment was observed by day +100 for all 24 participants. Of the 3 patients in the 75-mg dose cohort, 1 patient experienced neutrophil engraftment at day 15, and the remaining 2 patients engrafted at day 22. In the 300-mg dose cohort, the median (interquartile range) time to neutrophil engraftment was 14 (range 13 to 17) days.
TEAEs
All participants experienced at least 1 TEAE (Table 2). TEAEs were considered related to vedolizumab in 8 of 24 participants (33.3%; n = 2 and n = 6 in the 75-mg and 300-mg dose cohorts, respectively) (Table 2). Serious TEAEs were observed in 13 of 24 participants (54.2%) (36 events [4 in the 75-mg dose cohort and 32 in the 300-mg dose cohort]), including 1 participant in the 300-mg dose cohort who experienced 2 serious TEAEs: febrile neutropenia and hypotension. All TEAEs considered related to vedolizumab are shown in Table 3. No TEAEs led to discontinuation from the study or from vedolizumab therapy. The frequencies of all serious TEAEs and the most frequent TEAEs are provided in supplemental Tables 3 and 4, respectively. In the 300-mg dose cohort, the most common infections were cytomegalovirus (CMV)-related (7 participants in the 300-mg dose cohort, 6 of whom had CMV reactivation/viremia, and 1 CMV colitis) and Clostridium difficile–related infections (1 participant in the 75-mg dose cohort and 4 participants in the 300-mg dose cohort) (supplemental Table 4). C difficile–related infection occurred on the same day as the first dose of vedolizumab in 1 participant and in the context of broad-spectrum antibiotic exposure for the other 3 participants. During the treatment-emergent period, a total of 3 deaths was reported (Table 2), none of which were considered related to vedolizumab. One death was due to relapse of AML in a participant in the 75-mg dose cohort, and 2 deaths in the 300-mg dose cohort were due to aGVHD and relapse of ALL. Beyond the treatment-emergent period, an additional death related to relapse of AML was recorded; therefore, a total of 4 deaths was observed (see details of overall survival in “Efficacy”).
Overview of safety events in the study sample, by dose cohort
| Category of safety event | Vedolizumab 75 mg (n = 3) | Vedolizumab 300 mg (n = 21) | Total (N = 24) |
|---|---|---|---|
| TEAEs* | |||
| Total | 3 (100) | 21 (100) | 24 (100) |
| Related to study drug | 2 (66.7) | 6 (28.6) | 8 (33.3) |
| Not related to study drug | 1 (33.3) | 15 (71.4) | 16 (66.7) |
| Grade III or higher† | 3 (100) | 21 (100) | 24 (100) |
| Grade III or higher drug related† | 0 (0) | 2 (9.5) | 2 (8.3) |
| Leading to study drug discontinuation | 0 (0) | 0 (0) | 0 (0) |
| Serious AEs | |||
| Total | 2 (66.7) | 11 (52.4) | 13 (54.2) |
| Related to study drug | 0 (0) | 1 (4.8) | 1 (4.2) |
| Not related to study drug | 2 (66.7) | 10 (47.6) | 12 (50.0) |
| Leading to study drug discontinuation | 0 (0) | 0 (0) | 0 (0) |
| Relapse‡ | 1 (33.3) | 1 (4.8) | 2 (8.3) |
| Death‡ | 1 (33.3) | 2 (9.5) | 3 (12.5) |
| Category of safety event | Vedolizumab 75 mg (n = 3) | Vedolizumab 300 mg (n = 21) | Total (N = 24) |
|---|---|---|---|
| TEAEs* | |||
| Total | 3 (100) | 21 (100) | 24 (100) |
| Related to study drug | 2 (66.7) | 6 (28.6) | 8 (33.3) |
| Not related to study drug | 1 (33.3) | 15 (71.4) | 16 (66.7) |
| Grade III or higher† | 3 (100) | 21 (100) | 24 (100) |
| Grade III or higher drug related† | 0 (0) | 2 (9.5) | 2 (8.3) |
| Leading to study drug discontinuation | 0 (0) | 0 (0) | 0 (0) |
| Serious AEs | |||
| Total | 2 (66.7) | 11 (52.4) | 13 (54.2) |
| Related to study drug | 0 (0) | 1 (4.8) | 1 (4.2) |
| Not related to study drug | 2 (66.7) | 10 (47.6) | 12 (50.0) |
| Leading to study drug discontinuation | 0 (0) | 0 (0) | 0 (0) |
| Relapse‡ | 1 (33.3) | 1 (4.8) | 2 (8.3) |
| Death‡ | 1 (33.3) | 2 (9.5) | 3 (12.5) |
Data are presented as n (%).
A TEAE was defined as any AE that occurs after administration of the first dose of study drug and up to 18 weeks after the last dose of study medication.
Intensity for each AE was determined using the Common Terminology Criteria for Adverse Events, Version 4.03.22
Deaths and relapses were recorded within 18 weeks after the last dose. An additional participant relapsed and died after the 18-week treatment-emergent period.
Frequency of drug-related TEAEs, by dose cohort and system organ class
| Category of event* | Vedolizumab 75 mg (n = 3) | Vedolizumab 300 mg (n = 21) | Total (N = 24) |
|---|---|---|---|
| Participants with any drug-related TEAE† | 2 (66.7) | 6 (28.6) | 8 (33.3) |
| Blood and lymphatic system disorders | |||
| Total | 0 (0) | 2 (9.5) | 2 (8.3) |
| Anemia | 0 (0) | 1 (4.8) | 1 (4.2) |
| Febrile neutropenia | 0 (0) | 1 (4.8) | 1 (4.2) |
| Nervous system disorders | |||
| Total | 0 (0) | 2 (9.5) | 2 (8.3) |
| Headache | 0 (0) | 1 (4.8) | 1 (4.2) |
| Cognitive disorder | 0 (0) | 1 (4.8) | 1 (4.2) |
| Musculoskeletal and connective tissue disorders | |||
| Total | 1 (33.3) | 1 (4.8) | 2 (8.3) |
| Arthralgia | 0 (0) | 1 (4.8) | 1 (4.2) |
| Musculoskeletal pain | 1 (33.3) | 0 (0) | 1 (4.2) |
| Investigations | |||
| Total | 1 (33.3) | 1 (4.8) | 2 (8.3) |
| Alanine aminotransferase increased | 1 (33.3) | 0 (0) | 1 (4.2) |
| Aspartate aminotransferase increased | 0 (0) | 1 (4.8) | 1 (4.2) |
| Infections and infestations | |||
| CMV infection‡ | 0 (0) | 1 (4.8) | 1 (4.2) |
| Vascular disorders | |||
| Hypotension | 0 (0) | 1 (4.8) | 1 (4.2) |
| Respiratory, thoracic, and mediastinal disorders | |||
| Dyspnea exertional | 0 (0) | 1 (4.8) | 1 (4.2) |
| Skin and subcutaneous tissue disorders | |||
| Rash pruritic | 0 (0) | 1 (4.8) | 1 (4.2) |
| General disorders and administration site conditions | |||
| Pyrexia | 0 (0) | 1 (4.8) | 1 (4.2) |
| Participants with at least 1 serious drug-related TEAE | 0 (0) | 1 (4.8) | 1 (4.2) |
| Blood and lymphatic system disorders | |||
| Febrile neutropenia | 0 (0) | 1 (4.8) | 1 (4.2) |
| Vascular disorders | |||
| Hypotension | 0 (0) | 1 (4.8) | 1 (4.2) |
| Category of event* | Vedolizumab 75 mg (n = 3) | Vedolizumab 300 mg (n = 21) | Total (N = 24) |
|---|---|---|---|
| Participants with any drug-related TEAE† | 2 (66.7) | 6 (28.6) | 8 (33.3) |
| Blood and lymphatic system disorders | |||
| Total | 0 (0) | 2 (9.5) | 2 (8.3) |
| Anemia | 0 (0) | 1 (4.8) | 1 (4.2) |
| Febrile neutropenia | 0 (0) | 1 (4.8) | 1 (4.2) |
| Nervous system disorders | |||
| Total | 0 (0) | 2 (9.5) | 2 (8.3) |
| Headache | 0 (0) | 1 (4.8) | 1 (4.2) |
| Cognitive disorder | 0 (0) | 1 (4.8) | 1 (4.2) |
| Musculoskeletal and connective tissue disorders | |||
| Total | 1 (33.3) | 1 (4.8) | 2 (8.3) |
| Arthralgia | 0 (0) | 1 (4.8) | 1 (4.2) |
| Musculoskeletal pain | 1 (33.3) | 0 (0) | 1 (4.2) |
| Investigations | |||
| Total | 1 (33.3) | 1 (4.8) | 2 (8.3) |
| Alanine aminotransferase increased | 1 (33.3) | 0 (0) | 1 (4.2) |
| Aspartate aminotransferase increased | 0 (0) | 1 (4.8) | 1 (4.2) |
| Infections and infestations | |||
| CMV infection‡ | 0 (0) | 1 (4.8) | 1 (4.2) |
| Vascular disorders | |||
| Hypotension | 0 (0) | 1 (4.8) | 1 (4.2) |
| Respiratory, thoracic, and mediastinal disorders | |||
| Dyspnea exertional | 0 (0) | 1 (4.8) | 1 (4.2) |
| Skin and subcutaneous tissue disorders | |||
| Rash pruritic | 0 (0) | 1 (4.8) | 1 (4.2) |
| General disorders and administration site conditions | |||
| Pyrexia | 0 (0) | 1 (4.8) | 1 (4.2) |
| Participants with at least 1 serious drug-related TEAE | 0 (0) | 1 (4.8) | 1 (4.2) |
| Blood and lymphatic system disorders | |||
| Febrile neutropenia | 0 (0) | 1 (4.8) | 1 (4.2) |
| Vascular disorders | |||
| Hypotension | 0 (0) | 1 (4.8) | 1 (4.2) |
Data are presented as n (%).
MedDRA (version 21.0) terms were used for coding AEs.
Participants with 1 or more events within a level of MedDRA term are counted only once in that level.
Investigator reported this event as “CMV reactivation.”
Pharmacokinetics and immunogenicity
Vedolizumab was administered on days −1, +13, and +42, and serum concentration of vedolizumab increased from day −1 to day +13, plateauing at day +42. Serum vedolizumab concentration was increased in the 300-mg dose cohort compared with the 75-mg dose cohort, to an extent that was approximately proportional to the dose and followed a similar profile to the 75-mg dose cohort (Figure 1). Cmax was ∼1.8-fold higher on day 42 compared with day −1 following administration of both 75-mg and 300-mg doses, reflecting accumulation of vedolizumab after repeat dosing. A summary of serum vedolizumab PK parameters (AUC, Cmax, and Ctrough) by dose level is reported in Table 4. Interparticipant variability for both dose cohorts, as measured by the coefficient of variation (geometric CV), ranged from 23% to 69% for AUC and 26% to 56% for Cmax. Geometric mean Ctrough was 5.34 µg/mL and 20.65 µg/mL on day +13, and 7.01 µg/mL and 26.48 µg/mL on day +42, in the 75-mg and 300-mg dose cohorts, respectively (Table 4). PK results at the 300-mg dose level were considered sufficient for target saturation, such that further dose escalation was not required. No participants had detectable AVAs.
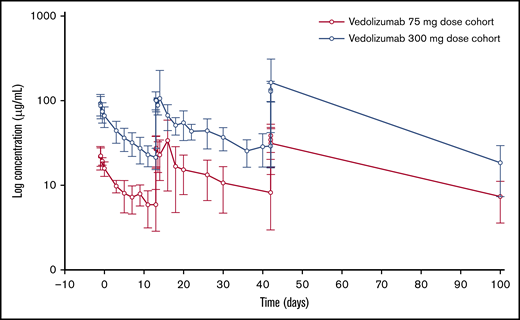
Summary of PK parameters of vedolizumab
| Day relative to allo-HSCT | Dose level, mg | Parameter | AUC, d × µg/mL | Cmax, µg/mL | Ctrough, µg/mL |
|---|---|---|---|---|---|
| Day −1 | 75 | n | 3 | 3 | NA |
| Mean (SD) | 136.9 (39.8) | 22.6 (6.2) | |||
| CV, % | 29 | 28 | |||
| Geometric mean | 133.1 | 22.1 | |||
| Geometric CV, % | 30 | 26 | |||
| 300 | n | 21 | 21 | ||
| Mean (SD) | 540.8 (142.5) | 92.2 (26.1) | |||
| CV | 26 | 28 | |||
| Geometric mean | 526.1 | 89.0 | |||
| Geometric CV | 23 | 27 | |||
| Day +13 | 75 | n | 3 | 3 | 3 |
| Mean (SD) | 390.7 (234.3) | 31.8 (17.4) | 5.9 (3.0) | ||
| CV | 60 | 55 | 51 | ||
| Geometric mean | 344.1 | 28.9 | 5.34 | ||
| Geometric CV | 69 | 56 | 58 | ||
| 300 | n | 19 | 20 | 21 | |
| Mean (SD) | 1469.5 (612.6) | 132.8 (116.7) | 21.3 (6.1) | ||
| CV | 42 | 88 | 28 | ||
| Geometric mean | 1373.3 | 114.3 | 20.7 | ||
| Geometric CV | 38 | 48 | 26 | ||
| Day +42 | 75 | n | 2 | 3 | 3 |
| Mean (SD) | 1326.5 (748.9) | 40.2 (13.7) | 8.2 (5.2) | ||
| CV | 56 | 34 | 64 | ||
| Geometric mean | 1216.2 | 38.4 | 7.0 | ||
| Geometric CV | 66 | 40 | 80 | ||
| 300 | n | 17 | 19 | 19 | |
| Mean (SD) | 5350.9 (4404.8) | 169.7 (140.6) | 29.1 (13.0) | ||
| CV | 82 | 83 | 45 | ||
| Geometric mean | 4545.7 | 147.2 | 26.5 | ||
| Geometric CV | 55 | 48 | 47 |
| Day relative to allo-HSCT | Dose level, mg | Parameter | AUC, d × µg/mL | Cmax, µg/mL | Ctrough, µg/mL |
|---|---|---|---|---|---|
| Day −1 | 75 | n | 3 | 3 | NA |
| Mean (SD) | 136.9 (39.8) | 22.6 (6.2) | |||
| CV, % | 29 | 28 | |||
| Geometric mean | 133.1 | 22.1 | |||
| Geometric CV, % | 30 | 26 | |||
| 300 | n | 21 | 21 | ||
| Mean (SD) | 540.8 (142.5) | 92.2 (26.1) | |||
| CV | 26 | 28 | |||
| Geometric mean | 526.1 | 89.0 | |||
| Geometric CV | 23 | 27 | |||
| Day +13 | 75 | n | 3 | 3 | 3 |
| Mean (SD) | 390.7 (234.3) | 31.8 (17.4) | 5.9 (3.0) | ||
| CV | 60 | 55 | 51 | ||
| Geometric mean | 344.1 | 28.9 | 5.34 | ||
| Geometric CV | 69 | 56 | 58 | ||
| 300 | n | 19 | 20 | 21 | |
| Mean (SD) | 1469.5 (612.6) | 132.8 (116.7) | 21.3 (6.1) | ||
| CV | 42 | 88 | 28 | ||
| Geometric mean | 1373.3 | 114.3 | 20.7 | ||
| Geometric CV | 38 | 48 | 26 | ||
| Day +42 | 75 | n | 2 | 3 | 3 |
| Mean (SD) | 1326.5 (748.9) | 40.2 (13.7) | 8.2 (5.2) | ||
| CV | 56 | 34 | 64 | ||
| Geometric mean | 1216.2 | 38.4 | 7.0 | ||
| Geometric CV | 66 | 40 | 80 | ||
| 300 | n | 17 | 19 | 19 | |
| Mean (SD) | 5350.9 (4404.8) | 169.7 (140.6) | 29.1 (13.0) | ||
| CV | 82 | 83 | 45 | ||
| Geometric mean | 4545.7 | 147.2 | 26.5 | ||
| Geometric CV | 55 | 48 | 47 |
Anomalous concentrations are excluded.
HSCT, hematopoietic stem cell transplantation; NA, not applicable.
Efficacy
aGVHD.
No participants in the 75-mg dose cohort (n = 3) developed modified Glucksberg grade II to IV aGVHD by 100 days after allo-HSCT. Of the 21 participants in the 300-mg dose cohort, only 4 participants (19%) developed overall grade II to IV aGVHD by 100 days after allo-HSCT; 3 participants developed grade II aGVHD, and 1 participant developed grade III aGVHD. No cases of grade IV aGVHD were observed. Of these 4 participants, 3 in the 300-mg dose cohort developed stage 1 aGVHD of the lower-intestinal tract, and no participants developed stages 2 to 4 lower-intestinal aGVHD. The participant with grade III aGVHD had stage 3 liver involvement (Table 5).
Summary of aGVHD by 100 days after allo-HSCT and 12 months after allo-HSCT, by grade and organ involvement
| aGVHD after allo-HSCT | Vedolizumab 75 mg (n = 3), n | Vedolizumab 300 mg (n = 21), n (%)/95% CI* |
|---|---|---|
| By 100 d | ||
| Frequency of aGVHD by grade | ||
| Grade II | 0 | 3 (14.3)/3.8, 41.1 |
| Grade III | 0 | 1 (4.8)/0.6, 29.7 |
| Grade IV | 0 | 0 |
| Frequency of aGVHD by affected organ involvement† | ||
| Skin only | 0 | 1 (4.8) |
| Skin and intestinal tract | 0 | 2 (9.5) |
| Skin, intestinal tract, and liver | 0 | 1 (4.8) |
| Frequency of lower-intestinal aGVHD by stage | ||
| Stage 1 | 0 | 3 (14.3)/4.3, 38.0 |
| Stages 2 to 4 | 0 | 0 |
| By 12 mo | ||
| Frequency of aGVHD by grade | ||
| Grade II | 0 | 3 (14.3)/3.8, 41.1 |
| Grade III | 0 | 2 (9.5)/2.0, 35.6 |
| Grade IV | 0 | 0 |
| Frequency of aGVHD by affected organ involvement† | ||
| Skin only | 0 | 2 (9.5) |
| Skin and intestinal tract | 0 | 2 (9.5) |
| Skin, intestinal tract, and liver | 0 | 1 (4.8) |
| Frequency of lower-intestinal aGVHD by stage | ||
| Stage 1 | 0 | 2 (9.5)/2.1, 34.3 |
| Stage 2 | 0 | 1 (4.8)/0.6, 28.3 |
| Stages 3 to 4 | 0 | 0 |
| aGVHD after allo-HSCT | Vedolizumab 75 mg (n = 3), n | Vedolizumab 300 mg (n = 21), n (%)/95% CI* |
|---|---|---|
| By 100 d | ||
| Frequency of aGVHD by grade | ||
| Grade II | 0 | 3 (14.3)/3.8, 41.1 |
| Grade III | 0 | 1 (4.8)/0.6, 29.7 |
| Grade IV | 0 | 0 |
| Frequency of aGVHD by affected organ involvement† | ||
| Skin only | 0 | 1 (4.8) |
| Skin and intestinal tract | 0 | 2 (9.5) |
| Skin, intestinal tract, and liver | 0 | 1 (4.8) |
| Frequency of lower-intestinal aGVHD by stage | ||
| Stage 1 | 0 | 3 (14.3)/4.3, 38.0 |
| Stages 2 to 4 | 0 | 0 |
| By 12 mo | ||
| Frequency of aGVHD by grade | ||
| Grade II | 0 | 3 (14.3)/3.8, 41.1 |
| Grade III | 0 | 2 (9.5)/2.0, 35.6 |
| Grade IV | 0 | 0 |
| Frequency of aGVHD by affected organ involvement† | ||
| Skin only | 0 | 2 (9.5) |
| Skin and intestinal tract | 0 | 2 (9.5) |
| Skin, intestinal tract, and liver | 0 | 1 (4.8) |
| Frequency of lower-intestinal aGVHD by stage | ||
| Stage 1 | 0 | 2 (9.5)/2.1, 34.3 |
| Stage 2 | 0 | 1 (4.8)/0.6, 28.3 |
| Stages 3 to 4 | 0 | 0 |
95% confidence intervals (CIs) were computed for the cumulative incidence of aGVHD by grade and were constructed using simultaneous CIs for multinomial proportions.37
Frequency of aGVHD by affected organ involvement is reported only for events of grade II to IV.
Grades of aGVHD are based on the modified Glucksberg criteria.
At 12 months after allo-HSCT, no participants in the 75-mg dose cohort (n = 3) had developed grade II to IV aGVHD. In the 300-mg dose cohort at 12 months, 3 participants (14.3%) developed grade II aGVHD; 2 participants (9.5%) developed grade III aGVHD, and no participants developed grade IV aGVHD. By 12 months, no additional participants had developed aGVHD of the lower-intestinal tract. One of the participants who had stage 1 lower-intestinal disease in the first 100 days experienced a recurrence of symptoms that progressed to stage 2 (Table 5).
In the 300-mg dose cohort, the Kaplan-Meier estimate of grade II to IV aGVHD-free survival probability by 12 months was 63.2% (95% CI, 36.7% to 81.1%) and grade III to IV aGVHD-free survival probability by 12 months was 79.6%.
Relapse, survival, and NRM.
By 12 months after allo-HSCT, 1 participant experienced underlying disease relapse in the 75-mg dose cohort (n = 3), and 2 participants experienced disease relapse in the 300-mg dose cohort (n = 21). There were 4 deaths in total, including 1 death in the 300-mg dose cohort that occurred beyond the 18-week treatment-emergent period with no deaths considered related to the study drug. Causes of death included disease relapse (n = 3) and aGVHD (n = 1). In the 300-mg dose cohort, overall survival at 12 months was 84.7% (Figure 2A), and NRM at 12 months was 5.6% (Figure 2B).

Overall survival and NRM at 12 months by dose cohort. (A) Overall survival. (B) NRM.
Overall survival and NRM at 12 months by dose cohort. (A) Overall survival. (B) NRM.
Discussion
Our findings demonstrate that 300 mg IV of vedolizumab is well tolerated when added to tacrolimus and MTX. Impressively, participants receiving 300 mg IV of vedolizumab experienced a relatively low incidence of lower-intestinal aGVHD in this small study. No DLTs were observed in any of the 24 participants; specifically, there was no delay or failure of hematopoietic engraftment, and no safety signals attributable to vedolizumab were identified. One participant in the 300-mg dose cohort experienced 2 serious TEAEs (febrile neutropenia and hypotension) possibly related to vedolizumab. In addition, PK data demonstrated that vedolizumab IV at a dose of 300 mg is expected to maintain α4β7 integrin receptor saturation over the dosing interval.23
Acute GVHD involving the lower-intestinal tract, which is often resistant to therapy and therefore responsible for the majority of morbidity and mortality associated with aGVHD,8,26,27 developed in only 3 participants in the 300-mg dose cohort by 12 months. In the 100 days after allo-HSCT, only stage 1 intestinal aGVHD event was observed for 3 participants. For 1 participant, recurrent intestinal aGVHD after 100 days was classified as stage 2 but had completely resolved by the last study visit. No participant developed stage 3 to 4 intestinal aGVHD, which is associated with significant mortality (overall survival, 25%).28 In previous large studies of patients undergoing allo-HSCT with calcineurin inhibitor (CNI)-based GVHD prophylaxis, the reported cumulative incidence of stage 2 to 4 lower-intestinal aGVHD was between 15% and 26%.5,7
Effective prevention of aGVHD, especially lower-intestinal aGVHD, has been an important goal for physicians treating patients undergoing allo-HSCT.3 Over the past 3 decades, there has been progress in reducing the severity, overall incidence, and prognosis of aGVHD.6 However, these improvements have likely been driven by improvements in HLA typing, donor and patient selection, and advancements in supportive care, rather than actual innovation in agents used to directly prevent aGVHD. Building on a standard backbone of CNI-based therapy, substitution of MTX with mycophenolate mofetil29 did not result in any clear benefit in previous studies. Similarly, recent attempts to add agents to the CNI/MTX combination using either bortezomib or maraviroc did not suggest any additional benefit in prevention of lower gastrointestinal aGVHD.30 Historically, many centers have added polyclonal anti–T-cell antibodies (most commonly, thymoglobulin or anti–T-lymphocyte globulin) to CNI/MTX, especially when using unrelated donors. Although there appears to be a reduction in chronic GVHD with anti–T-cell antibody products, the benefit in terms of a decrease in aGVHD is inconsistent,31-33 and a recent phase 3 randomized trial in the United States suggested increased mortality with the addition of anti–T-lymphocyte globulin compared with standard CNI/MTX GVHD prophylaxis.34 Most recently, 2 additional modalities of GVHD prevention, specifically posttransplant high-dose cyclophosphamide-based regimens and ex vivo T-cell depletion, are being compared with standard CNI/MTX in ongoing prospective clinical trials. Although we await the results of such trials, prevention of intestinal aGVHD remains a significant unmet need for allo-HSCT recipients.
Vedolizumab specifically binds to α4β7 integrin and inhibits adhesion to MAdCAM-1, unlike other integrin inhibitors such as pan-α4 integrin inhibitors that bind to both α4β1 and α4β7, which then inhibits cellular adhesion to VCAM-1 and MAdCAM-1, respectively. The α4β7 integrin is highly expressed on a subset of memory T-helper lymphocytes that migrate preferentially into the intestinal tract, and MAdCAM-1 is mainly expressed on gut endothelial cells and is critical in the homing of these T lymphocytes to the intestinal tract. Vedolizumab, unlike other novel therapies studied for GVHD prophylaxis, exerts a gut-selective immunomodulatory mechanism of action. In nonclinical and clinical studies in healthy human volunteers, vedolizumab did not interfere with the systemic immune response.35,36 This is a potential mechanistic advantage over other GVHD prophylaxis therapies where additive systemic immunosuppression may pose substantial risk, such as an increased risk of infections or impairment of the therapeutic graft versus malignancy effect. The findings of the current study have shown no early signal that vedolizumab adversely affects the rate of infection or relapse. Limitations of this study include the small sample size and the heterogeneous clinical characteristics, including different donor stem cell sources and conditioning regimen intensities.
In conclusion, vedolizumab administered on days −1, +13, and +42 in relation to allo-HSCT was well tolerated when added to standard GVHD prophylaxis. There was no effect on engraftment, and no safety concerns were observed. The low cumulative incidence of lower-intestinal aGVHD and overall grade III to IV aGVHD are compelling. The results from this study support further evaluation of vedolizumab IV 300 mg added to standard GVHD prophylaxis in patients undergoing allo-HSCT, and a phase 3 randomized study (#NCT03657160) is ongoing.
Takeda makes patient-level, deidentified data sets, and associated documents available after applicable marketing approvals and commercial availability have been received, an opportunity for the primary publication of the research has been allowed, and other criteria have been met as set forth in Takeda’s Data Sharing Policy (www.takedaclinicaltrials.com). To obtain access, researchers must submit a legitimate academic research proposal for adjudication by an independent review panel, who will review the scientific merit of the research and the requestor’s qualifications and conflict of interest that can result in potential bias. Once approved, qualified researchers who sign a data sharing agreement are provided access to these data in a secure research environment.
Acknowledgments
This work was supported by Takeda Pharmaceutical Company, Ltd. Lisa Law (ORCID iD: https://orcid.org/0000-0002-9837-6609) of Oxford PharmaGenesis (Oxford, United Kingdom) provided medical writing support, which was funded by Takeda Pharmaceutical Company, Ltd, in accordance with Good Publication Practice 3 (GPP3).
Authorship
Contribution: All authors contributed to the conception, design, and analysis/interpretation of this study; contributed to the drafting of the paper or to revising it critically for intellectual content; and gave final approval of the version to be published and agree to be accountable for all aspects of the work.
Conflict-of-interest disclosure: Y.-B.C. has received consultancy fees for AbbVie, Incyte, Kiadis Pharma, Magenta Therapeutics, and Takeda Pharmaceuticals. N.N.S. is on advisory boards for Celgene, Cellectar Biosciences, Juno Pharmaceuticals, and Kite; has received research funding from BMS and Miltenyi Biotec; has received travel support from Miltenyi Biotec; and has received consultancy fees from Incyte and Celgene. C. Cutler has received consultancy fees from Incyte, Jazz Pharmaceuticals, Bristol-Myers Squibb, Genentech, and Pharmacyclics. J.J. is employed by Takeda Pharmaceuticals International. M.A. is an employee of Takeda. C. Chen is an employee of Takeda Development Center Americas, Inc. S.Q. is employed by Takeda Pharmaceuticals International. A.P. was formally employed by Takeda Pharmaceutical Company Ltd. The remaining authors declare no competing financial interests.
Correspondence: Yi-Bin Chen, Massachusetts General Hospital, Zero Emerson Pl, Suite 118, Boston, MA 02114; e-mail: ychen6@partners.org.

