

Key Points
Vascular SMC CYB5R3 delays development of SCD-associated PH in mice.
PH in SCD can exist in mice by 5 weeks of age when SMC CYB5R3 protein is deficient.

Abstract
Pulmonary and systemic vasculopathies are significant risk factors for early morbidity and death in patients with sickle cell disease (SCD). An underlying mechanism of SCD vasculopathy is vascular smooth muscle (VSM) nitric oxide (NO) resistance, which is mediated by NO scavenging reactions with plasma hemoglobin (Hb) and reactive oxygen species that can oxidize soluble guanylyl cyclase (sGC), the NO receptor. Prior studies show that cytochrome b5 reductase 3 (CYB5R3), known as methemoglobin reductase in erythrocytes, functions in VSM as an sGC heme iron reductase critical for reducing and sensitizing sGC to NO and generating cyclic guanosine monophosphate for vasodilation. Therefore, we hypothesized that VSM CYB5R3 deficiency accelerates development of pulmonary hypertension (PH) in SCD. Bone marrow transplant was used to create SCD chimeric mice with background smooth muscle cell (SMC)–specific tamoxifen-inducible Cyb5r3 knockout (SMC R3 KO) and wild-type (WT) control. Three weeks after completing tamoxifen treatment, we observed 60% knockdown of pulmonary arterial SMC CYB5R3, 5 to 6 mm Hg elevated right-ventricular (RV) maximum systolic pressure (RVmaxSP) and biventricular hypertrophy in SS chimeras with SMC R3 KO (SS/R3KD) relative to WT (SS/R3WT). RV contractility, heart rate, hematological parameters, and cell-free Hb were similar between groups. When identically generated SS/R3 chimeras were studied 12 weeks after completing tamoxifen treatment, RVmaxSP in SS/R3KD had not increased further, but RV hypertrophy relative to SS/R3WT persisted. These are the first studies to establish involvement of SMC CYB5R3 in SCD-associated development of PH, which can exist in mice by 5 weeks of SMC CYB5R3 protein deficiency.
Introduction
We have appreciated for more than 50 years that a basic protein abnormality in the hemoglobin (Hb) molecule underlies sickle cell disease (SCD) pathogenesis. Polymerization of intraerythrocytic mutant Hb (HbS) under low oxygen and/or low pH conditions ultimately leads to recurring transient episodes of vascular obstruction similar to ischemia-reperfusion injury with its associated oxidative stress.1-3 Chronic intravascular hemolysis also generates oxidative stress by Fenton chemistry and nitric oxide (NO) scavenging.4-8 As patients with SCD age, these toxic effects promote progressive pulmonary and systemic vasculopathy, contributing to the multiple, overlapping causes of systemic and pulmonary hypertension (PH) development.9,10 This is mechanistically defined in patients by increased pulmonary vascular resistance, elevated pulse pressure, vascular stiffness, endothelial dysfunction, and intimal/medial proliferation.11-18
Definitively diagnosed by right-heart catheterization, PH occurs in 6% to 11% of adults with SCD and independently predicts early death.16-18 Approximately one-half of PH patients have pulmonary arterial hypertension (PAH) and one-half have pulmonary venous hypertension,16,17,19 which arise on opposite ends of the pulmonary vasculature, precapillary and postcapillary, respectively. PH in SCD, whether precapillary or postcapillary, is often associated with “nitric oxide resistance,” characterized by the inability of the NO donor, sodium nitroprusside, to vasodilate.4,6,8 Although these pathological effects typically correlate with levels of plasma Hb, endothelial dysfunction, increasing pulmonary pressures, and risk of death,11-13,15-18,20,21 compensatory and aggravating factors are incompletely understood. Furthermore, oxidation and inactivation of the NO target soluble guanylyl cyclase (sGC) may also underlie vascular resistance to NO in SCD.22
NO is the primary activator of sGC in vascular smooth muscle (VSM) for production of cyclic guanosine monophosphate (cGMP), which activates protein kinase G (PKG)–mediated vasodilation in mammalian organisms.23-25 The major prerequisite for NO binding and activation of sGC is reduced heme iron (Fe2+) in the sGC-active site. sGC heme iron oxidation (Fe3+) and subsequent loss of heme abolishes NO-induced, sGC-mediated cGMP production.26,27 Nicotinamide adenine dinucleotide (NADH) cytochrome b5 (Cyb5) reductase 3 (CYB5R3) is a flavoprotein that transfers electrons from NADH through Cyb5 to an electron acceptor. We have previously shown in rat aortic smooth muscle cell (SMC) that CYB5R3 transfers electrons to sGC heme iron, recycling it from the oxidized (Fe3+) to reduced (Fe2+) form for NO responsiveness.28 Moreover, we recently found that loss of SMC CYB5R3 caused a modest 5.84 mm Hg increase in systemic blood pressure, which increased to 14.75 mm Hg in mice challenged with angiotensin II.29 This suggests that without sGC oxidation, the reducing role of Cyb5R3 in sGC’s vasorelaxation activity should be minimal and hence undetectable by right-ventricular (RV) maximum systolic pressure (RVmaxSP) measurements. NO resistance in SCD may stem from deficient sGC redox regulation by CYB5R3.
To test our hypothesis that development of PH is accelerated in SCD when SMC-specific CYB5R3 is not available for sGC redox regulation, we used SS-transgenic animals, which have enhanced oxidative stress relative to their AS and AA counterparts.30-32
For roughly 45 years, starting in 1973 with recommendations coming out of the 1st World Symposium on Pulmonary Hypertension, precapillary PH was considered definitively diagnosed when a mean pulmonary artery pressure (PAP; mPAP), measured by right-heart catheterization, was ≥25 mm Hg. Given a normal resting mPAP of 14.0 ± 3.3 mm Hg, the mPAP threshold for diagnosing all precapillary PH was recently revised to >20 mm Hg, with the recommendation for best diagnosis including concomitant pulmonary arterial wedge pressure (PAWP) <15 mm Hg and pulmonary vascular resistance (PVR) ≥3 Wood units (WU).33 We report accelerated, spontaneous development of RVmaxSP exceeding 20 mm Hg and biventricular cardiac hypertrophy at 3 weeks postknockdown of SMC CYB5R3 in mice with SCD. The PH is accompanied by increased RV end diastolic pressure, but not enhanced anemia or cell-free Hb.
Materials and methods
Animals
C57BL/6J (stock # 000664) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Cyb5r3fl/flMyh11-CreERT2 mice (described in detail in supplemental Materials)22,29,34 and Townes knock‐in humanized sickle Hb (SS) mice and their humanized Hb controls (AA and AS) were bred on C57BL/6 and 129S backgrounds35 and maintained at the University of Pittsburgh. For the Cyb5r3fl/flMyh11-CreERT2 mice, the Myh11-driven Cre-ERT transgene resides on the Y chromosome,36 thus necessitating the use of male mice for all experiments, which were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Animals received adequate chow and drinking water and pathogen-free housing.
BM-transplanted chimeras
DNA analysis by polymerase chain reaction confirmed genetic identities. As labeled in text and figures, SS/R3KD orfl and AS/R3KD or fl chimeras are Cyb5r3fl/flMyh11-CreERT2 mice transplanted with Townes SS or AS bone marrow (BM), respectively. SS/R3WT or SS/R3wt and AS/R3WT or AS/R3wt chimeras are Cyb5rwt/wtMyh11-CreERT2 mice similarly transplanted. SS/R3fland AS/R3fl are pretamoxifen treatment; SS/R3KD and AS/R3KD are posttamoxifen treatment. At 8 to 10 weeks posttransplantation, Hb electrophoresis was performed using Hb (E) and Acid (E) Hb (E) kits (Sebia Capillary System, Lisse, France), following the manufacturer’s guidelines. Standard HbA, HbS, HbF, HbA2, and C57BL/6J blood were run as migration controls. Engraftment efficiency was determined by densitometry using Sebia system software (Sebia Phoresis rel. 8.6.3).37 Only chimeras engrafted ≥80% of donor Hb phenotype were used in subsequent experiments. SS/R3fl, AS/R3fl, SS/R3wt, and AS/R3wt chimeras received a tamoxifen diet (Teklad) for 2 weeks to activate Cre-loxp–mediated excision of exon 3 and loss of Cyb5r3 expression in Myh11+ cells.
Immunohistochemical analysis
Postmortem murine organs were weighed and formalin-fixed (Santa Cruz Biotechnology; 4% in phosphate-buffered saline [PBS]). Lungs were paraffin-embedded and sectioned (7 µM thickness). Sections were deparaffinized by dehydration in the following solutions for 5 minutes (2 times each): xylene, 100% ethanol, 95% ethanol, 70% ethanol, and double-distilled H2O. Sections were incubated in antigen-retrieval solution (0.9% in double-distilled H2O) and microwaved for 20 minutes. Sections were rehydrated in 1× PBS for 5 minutes. Blocking occurred in horse serum (10% in PBS, 1 hour at room temperature; Sigma-Aldrich), followed by incubation overnight (4°C) with primary antibodies CYB5R3 (1:100; Proteintech) and PECAM1 (1:250; Santa Cruz Biotechnology) in blocking buffer. Matching concentrations of rabbit (I-1000; Vector Laboratories) and goat immunoglobulin G controls (I-5000; Vector Laboratories) were applied to 1 section per slide. PBS-washed sections were incubated with preconjugated smooth muscle α-actin (ACTA2)–fluorescein isothiocyanate (clone 1A4, 1:500; Sigma-Aldrich), 4′,6-diamidino-2-phenylindole (DAPI) (1:100; ThermoFisher Scientific), and secondary antibodies donkey anti-rabbit AlexaFluor 596 (1:250; Invitrogen) and donkey anti-goat AlexaFluor 647 (1:250; Invitrogen). Prolong Gold Antifade with DAPI (Invitrogen) was used to coverslip sections.
Heart tissue was assessed for fibrosis using the Masson Trichrome kit (ThermoFisher), according to the manufacturer’s directions. Trichrome-stained sections were imaged using a TissueGnostics microscope with a 20× objective and stitched using NIS Elements software (Snake Stitch; 8% overlap). Individual images were converted into binary images, and trichrome-positive and total tissue area measured using Fiji software.
Closed-chest RV microcatheterization
Microcatheterization was performed as previously described.38 Briefly, mice were anesthetized with etomidate/urethane (9/1.1 mg/kg intraperitoneally) and body temperature (37°C) maintained by heating pad. The right neck was incised, exposing the external jugular vein for catheterization (1.2 F micropressure-volume). After 5 minutes of stabilization, pressure was recorded and saved for offline analysis using IOX2 software (EMKA Technologies, Falls Church, VA).
Hematological phenotyping
Complete blood counts (Heska; HemaTrue Inc, Miami Lakes, FL) were conducted in EDTA-anticoagulated blood (5% vol/vol) sampled (50 μL) in anesthetized (1% isoflurane) mice by retroorbital puncture. Plasma was centrifuge-separated (1500g for 10 minutes). Heme-containing species and their metabolites in plasma were determined by UV-Visible spectral deconvolution using the method of Oh et al.39
Statistical analysis
Results are presented as mean ± standard error of the mean (SEM). PRISM software (GraphPad version 7.0a for Mac; GraphPad Software, La Jolla, CA) was used to analyze response data. Hemodynamic response data between 2 groups was analyzed using the Student unpaired t test, with the Mann-Whitney U test for nonparametric analysis of non-Gaussian distributed data (F test). Hematological data within groups, pretamoxifen vs end-of study, were analyzed using the Student paired t test. Analysis of variance with the Holm-Sidak multiple comparisons test was used for analysis of response between 3 or more data groups. Correlation analyses used Pearson r. Statistical significance was set at P < .05.
Results
Generation of conditional tamoxifen-inducible smooth muscle–specific Cyb5r3 knockout mice
Cyb5r3 floxed animals (Cyb5r3fl/fl) were crossed with Myh11-CreERT2 mice36 to generate a tamoxifen-inducible SMC-specific CYB5R3 knockdown animal (supplemental Figure 1) for testing the role of SMC CYB5R3 protein in the development of SCD-associated PH. These Cyb5r3fl/flMyh11-CreERT2 animals were similar in appearance and behavior when compared with their wild-type (WT) counterparts (Cyb5r3wt/wtMyh11-CreERT2).
Effects of 3-week SMC CYB5R3 knockdown on RVmaxSP and RV remodeling in SS chimeras
Preliminary studies using WT C57BL/6J male mice as recipients of BM from Townes SS, AS, and AA mice informed the choice of BM transplantation protocol used for subsequently planned studies in the tamoxifen-inducible SMC-specific CYB5R3 knockdown mice. (supplemental Figure 2). These preliminary studies also showed similar hemodynamic phenotypes shared by the AA- and AS-transplanted C57BL/6J mice, neither of which developed PH after 14 weeks of ≥80% engraftment (supplemental Figure 2). Hence, AS donors were used to establish the humanized Hb control groups (AS/R3fl and AS/R3wt) for the SS/R3fl (Cyb5r3 floxed) and SS/R3wt (Cyb5r3 WT) BM-transplanted animals. At 8 to 10 weeks post-BM transplantation, hematological phenotyping was conducted and R3fl and R3wt chimeras demonstrating ≥80% conversion to donor Hb phenotype (HbS or HbAS) were placed on a 2-week tamoxifen diet to induce SMC-specific Cyb5r3 knockdown (SS/R3KD) (Figure 1). Chimeras were allowed an additional 3 weeks of aging on regular diet to determine effects of SMC CYB5R3 knockdown on PH development during early life with SCD (5 weeks total). Hematological analyses showed Hb, hematocrit, leukocyte, and platelet counts at end of study to be similar to pretamoxifen levels in the SS/R3KD group, indicating no effect for SMC CYB5R3 knockdown on blood composition (Table 1). RV catheterization revealed 5 to 6 mm Hg higher RVmaxSP in SS/R3KD (32.85 ± 2.06; n = 10) relative to SS/R3WT (24.36 ± 1.01; n = 13) (Figure 2A) with no difference in heart rate (Figure 2B). Both RV and left ventricular (LV) plus septum (LV+S) weights, normalized to tibia length (RV/tibia and LV+S/tibia, respectively), were also greater in SS/R3KD (1.67 ± 0.06 mg/mm and 5.58 ± 0.11 mg/mm; n = 14) relative to SS/R3WT (1.47 ± 0.05 mg/mm and 5.07 ± 0.18 mg/mm; n = 20) (Figure 2C-D), but no difference in Fulton Index was apparent. (supplemental Figure 3A). These biventricular enlargements in the CYB5R3 knockdown group occurred without apparent changes to contractile index (Figure 3A). In line with the higher RVmaxSP, mPAP and RV end diastolic pressure were also increased in SS/R3KD (22.02 ± 1.36 mm Hg and 3.003 ± 0.37 mm Hg; n = 10) relative to SS/R3WT (17.23 ± 1.01 mm Hg and 1.68 ± 0.32 mm Hg; n = 13) (Figure 3B-C). The combined results for 3-week knockdown of SMC CYB5R3 were unique to the SS phenotype as the corresponding AS-transplanted controls showed no apparent effects for 3-week SMC CYB5R3 knockdown on any hematological (Table 2) or hemodynamic parameters (Figure 4). The control data also indicate that 3 weeks of SMC CYB5R3 knockdown by itself does not induce spontaneous development of PH.
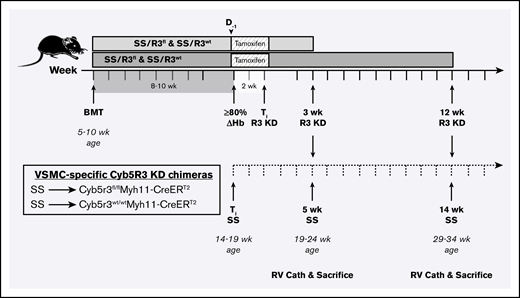
Study design for testing role of SMC CYB5R3 in SCD-associated PH.Cyb5r3fl/flMyh11-CreERT2 and Cyb5r3wt/wtMyh11-CreERT2 mice were lethally irradiated (500-550 rad twice) and transplanted with BM (2-4 million cells) from SS donor mice to create SS/R3fl and SS/R3wt chimeras, respectively. At 8 to 10 weeks posttransplantation, peripheral blood was sampled to test engraftment to donor Hb phenotype. Chimeras testing ≥80% engraftment, assessed by Hb electrophoresis, were fed a tamoxifen diet ad libitum for 2 weeks, and thereafter switched back to regular chow and aged for an additional 3 or 12 weeks before measurement of hemodynamics by RV microcatheterization. At 3 weeks CYB5R3 knockdown (solid timeline), animals were 5 weeks SS phenotype (dashed timeline) and a total age of 19 to 24 weeks. At 12 weeks CYB5R3 knockdown (solid timeline), animals were 14 weeks SS phenotype and a total age of 29 to 34 weeks. Complete blood counts were assessed at 8 to 10 weeks posttransplant, before a tamoxifen diet and again at time of RV catheterization. ≥80% ΔHb, time point at which animal is at least 80% engrafted to SS phenotype and used to calculate number of weeks with humanized HbS circulating red cells; BMT, BM transplant; D−1, 1 day before starting 2-week tamoxifen diet; RV Cath, right ventricle microcatheterization; Ti R3 KD, completion of tamoxifen treatment and initial time point of CYB5R3 knockdown.
Study design for testing role of SMC CYB5R3 in SCD-associated PH.Cyb5r3fl/flMyh11-CreERT2 and Cyb5r3wt/wtMyh11-CreERT2 mice were lethally irradiated (500-550 rad twice) and transplanted with BM (2-4 million cells) from SS donor mice to create SS/R3fl and SS/R3wt chimeras, respectively. At 8 to 10 weeks posttransplantation, peripheral blood was sampled to test engraftment to donor Hb phenotype. Chimeras testing ≥80% engraftment, assessed by Hb electrophoresis, were fed a tamoxifen diet ad libitum for 2 weeks, and thereafter switched back to regular chow and aged for an additional 3 or 12 weeks before measurement of hemodynamics by RV microcatheterization. At 3 weeks CYB5R3 knockdown (solid timeline), animals were 5 weeks SS phenotype (dashed timeline) and a total age of 19 to 24 weeks. At 12 weeks CYB5R3 knockdown (solid timeline), animals were 14 weeks SS phenotype and a total age of 29 to 34 weeks. Complete blood counts were assessed at 8 to 10 weeks posttransplant, before a tamoxifen diet and again at time of RV catheterization. ≥80% ΔHb, time point at which animal is at least 80% engrafted to SS phenotype and used to calculate number of weeks with humanized HbS circulating red cells; BMT, BM transplant; D−1, 1 day before starting 2-week tamoxifen diet; RV Cath, right ventricle microcatheterization; Ti R3 KD, completion of tamoxifen treatment and initial time point of CYB5R3 knockdown.
Blood cell indices for Townes SS BM–transplanted Cyb5R3wt and Cyb5R3fl chimeras before tamoxifen treatment (day −1) and at end of 3-week study
| Pretamoxifen | End of study | Statistical significance, P | ||||
|---|---|---|---|---|---|---|
| SS/R3wt, n = 19 | SS/R3fl, n = 14 | SS/R3WT, n = 19 | SS/R3KD, n = 14 | SS/R3WT vs SS/R3wt | SS/R3KD vs SS/R3fl | |
| Hct, % | 31.99 ± 0.49 | 32.95 ± 0.84 | 31.59 ± 0.59 | 33.61 ± 0.87 | .403 | .692 |
| Hgb, g/dL | 11.21 ± 0.18 | 11.56 ± 0.28 | 11.24 ± 0.16 | 11.59 ± 0.26 | .809 | .786 |
| MCV, fL | 47.67 ± 0.72 | 51.17 ± 0.93 | 49.62 ± 0.31 | 51.51 ± 0.77 | .007 | .092 |
| RDW, % | 35.09 ± 0.68 | 34.49 ± 0.61 | 35.69 ± 0.6 | 33.84 ± 0.81 | .091 | .089 |
| MCH, pg | 16.73 ± 0.27 | 17.32 ± 0.35 | 17.54 ± 0.1 | 17.85 ± 0.26 | .005 | .102 |
| RBC, 1012/L | 6.74 ± 0.15 | 6.51 ± 0.12 | 6.37 ± 0.12 | 6.49 ± 0.12 | .032 | .835 |
| WBC, ×109/L | 14.67 ± 0.8 | 14.08 ± 1.09 | 14.12 ± 0.71 | 13.4 ± 0.91 | .225 | .204 |
| PLT, ×109/L | 403 ± 13.3 | 353 ± 16.6 | 362 ± 13.5 | 335 ± 15.2 | .077 | .142 |
| MPV, fL | 6.6 ± 0.05 | 6.7 ± 0.08 | 6.8 ± 0.07 | 6.7 ± 0.07 | .025 | .095 |
| Pretamoxifen | End of study | Statistical significance, P | ||||
|---|---|---|---|---|---|---|
| SS/R3wt, n = 19 | SS/R3fl, n = 14 | SS/R3WT, n = 19 | SS/R3KD, n = 14 | SS/R3WT vs SS/R3wt | SS/R3KD vs SS/R3fl | |
| Hct, % | 31.99 ± 0.49 | 32.95 ± 0.84 | 31.59 ± 0.59 | 33.61 ± 0.87 | .403 | .692 |
| Hgb, g/dL | 11.21 ± 0.18 | 11.56 ± 0.28 | 11.24 ± 0.16 | 11.59 ± 0.26 | .809 | .786 |
| MCV, fL | 47.67 ± 0.72 | 51.17 ± 0.93 | 49.62 ± 0.31 | 51.51 ± 0.77 | .007 | .092 |
| RDW, % | 35.09 ± 0.68 | 34.49 ± 0.61 | 35.69 ± 0.6 | 33.84 ± 0.81 | .091 | .089 |
| MCH, pg | 16.73 ± 0.27 | 17.32 ± 0.35 | 17.54 ± 0.1 | 17.85 ± 0.26 | .005 | .102 |
| RBC, 1012/L | 6.74 ± 0.15 | 6.51 ± 0.12 | 6.37 ± 0.12 | 6.49 ± 0.12 | .032 | .835 |
| WBC, ×109/L | 14.67 ± 0.8 | 14.08 ± 1.09 | 14.12 ± 0.71 | 13.4 ± 0.91 | .225 | .204 |
| PLT, ×109/L | 403 ± 13.3 | 353 ± 16.6 | 362 ± 13.5 | 335 ± 15.2 | .077 | .142 |
| MPV, fL | 6.6 ± 0.05 | 6.7 ± 0.08 | 6.8 ± 0.07 | 6.7 ± 0.07 | .025 | .095 |
SS/R3wt and SS/R3fl are SS/R3WT and SS/R3KD groups, respectively, before tamoxifen treatment. Values are mean plus or minus SEM using the paired Student t test.
Hct, hematocrit; Hgb, total hemoglobin; MCH, mean corpuscular Hb; MCV, mean corpuscular volume; MPV, mean platelet volume; PLT, platelet; RBC, red blood cell; RDW, red blood cell distribution width; WBC, leukocyte.

Three weeks of SMC CYB5R3 knockdown accelerates development of PH in SS chimeras. Chimeras testing ≥80% engraftment, assessed by Hb electrophoresis, were aged an additional 3 weeks (A-D) or 12 weeks (E-H) on regular diet before measurement of hemodynamics by closed-chest RV microcatheterization. (A) At 3 weeks following completion of tamoxifen-induced KO, RVmaxSP for SS/R3KD (blue bar, n = 13) was increased relative to SS/R3WT (red bar, n = 10). (B) Heart rates (HR) between knockdown and WT groups were similar (n = 13 and 10, respectively). Both RV/tibia (C) and LV+S/tibia (D) were enlarged in the knockdown group (n = 20) relative to WT (n = 14) at the 3-week time point. For the 12-week study, no differences were observed between CYB5R3 knockdown and WT groups for RVmaxSP (E) and HR (F) (n = 5 and 6, respectively). (G) RV/tibia was enlarged in the knockdown group relative to WT, whereas (H) LV+S/tibia was similar between groups (n = 5 and 7, respectively). The mean ± SEM is represented. The Student unpaired t test was used to determine statistical significance. &A nonparametric Mann-Whitney U test was used to determine statistical significance when F test indicated a non-Gaussian distribution of variance between groups.
Three weeks of SMC CYB5R3 knockdown accelerates development of PH in SS chimeras. Chimeras testing ≥80% engraftment, assessed by Hb electrophoresis, were aged an additional 3 weeks (A-D) or 12 weeks (E-H) on regular diet before measurement of hemodynamics by closed-chest RV microcatheterization. (A) At 3 weeks following completion of tamoxifen-induced KO, RVmaxSP for SS/R3KD (blue bar, n = 13) was increased relative to SS/R3WT (red bar, n = 10). (B) Heart rates (HR) between knockdown and WT groups were similar (n = 13 and 10, respectively). Both RV/tibia (C) and LV+S/tibia (D) were enlarged in the knockdown group (n = 20) relative to WT (n = 14) at the 3-week time point. For the 12-week study, no differences were observed between CYB5R3 knockdown and WT groups for RVmaxSP (E) and HR (F) (n = 5 and 6, respectively). (G) RV/tibia was enlarged in the knockdown group relative to WT, whereas (H) LV+S/tibia was similar between groups (n = 5 and 7, respectively). The mean ± SEM is represented. The Student unpaired t test was used to determine statistical significance. &A nonparametric Mann-Whitney U test was used to determine statistical significance when F test indicated a non-Gaussian distribution of variance between groups.

Three weeks of SMC CYB5R3 knockdown increases mPAP and RV end diastolic pressure in SS chimeras. Chimeras testing 80% or greater engraftment, assessed by Hb electrophoresis, were aged an additional 3 weeks (A-F) or 12 weeks (G-L) on regular diet before measurement of hemodynamics by RV microcatheterization. RV contractile index (A), mPAP (B), RV end diastolic pressure (C), RV maximum pressure rise (dP/dT max) (D), τ Weiss (W) (E), and RV minimum pressure rise (dP/dT min) (F) for SS/R3KD (blue bar) and SS/R3WT chimeras (red bar) for the 3-week study. RV contractile index (G), mPAP (H), RV end diastolic pressure (I), RV dP/dT max (J), τ Weiss (W) (K), and RV dP/dT min (L) for SS/R3KD (blue bar) and SS/R3WT chimeras (red bar) in the 12-week study. RV Contractile Index was not affected by 3- (A) or 12-week (G) knockdown of CYB5R3 in smooth muscle. Differences between the mPAP (B) and RV end diastolic pressure (C) that had been apparent at 3-week knockdown of CYB5R3 in smooth muscle was no longer apparent at 12-week knockdown (panels H and I, respectively). The mean ± SEM is represented. A nonparametric Mann-Whitney U test was used to determine statistical significance when F test indicated a non-Gaussian distribution of variance between groups.
Three weeks of SMC CYB5R3 knockdown increases mPAP and RV end diastolic pressure in SS chimeras. Chimeras testing 80% or greater engraftment, assessed by Hb electrophoresis, were aged an additional 3 weeks (A-F) or 12 weeks (G-L) on regular diet before measurement of hemodynamics by RV microcatheterization. RV contractile index (A), mPAP (B), RV end diastolic pressure (C), RV maximum pressure rise (dP/dT max) (D), τ Weiss (W) (E), and RV minimum pressure rise (dP/dT min) (F) for SS/R3KD (blue bar) and SS/R3WT chimeras (red bar) for the 3-week study. RV contractile index (G), mPAP (H), RV end diastolic pressure (I), RV dP/dT max (J), τ Weiss (W) (K), and RV dP/dT min (L) for SS/R3KD (blue bar) and SS/R3WT chimeras (red bar) in the 12-week study. RV Contractile Index was not affected by 3- (A) or 12-week (G) knockdown of CYB5R3 in smooth muscle. Differences between the mPAP (B) and RV end diastolic pressure (C) that had been apparent at 3-week knockdown of CYB5R3 in smooth muscle was no longer apparent at 12-week knockdown (panels H and I, respectively). The mean ± SEM is represented. A nonparametric Mann-Whitney U test was used to determine statistical significance when F test indicated a non-Gaussian distribution of variance between groups.
Blood cell indices for Townes AS BM–transplanted Cyb5R3wt and Cyb5R3fl chimeras before tamoxifen treatment (day −1) and at end of 3-week study
| Pretamoxifen | End of study | Statistical significance, P | ||||
|---|---|---|---|---|---|---|
| AS/R3wt, n = 10 | AS/R3fl, n = 14 | AS/R3WT, n = 10 | AS/R3KD, n = 14 | AS/R3WT vs AS/R3wt | AS/R3KD vs AS/R3fl | |
| Hct, % | 36.74 ± 0.62 | 34.37 ± 0.69 | 35.19 ± 0.56 | 33.58 ± 0.58 | .079 | .389 |
| Hgb, g/dL | 13.08 ± 0.19 | 12.24 ± 0.21 | 12.74 ± 0.27 | 11.88 ± 0.16 | .323 | .188 |
| MCV, fL | 32.67 ± 0.5 | 32.3 ± 0.16 | 33.07 ± 0.68 | 33.16 ± 0.26 | .64 | .008 |
| RDW, % | 31.56 ± 1.23 | 29.99 ± 0.23 | 33.09 ± 1.84 | 31.51 ± 0.51 | .498 | .060 |
| MCH, pg | 11.66 ± 0.69 | 11.51 ± 0.07 | 11.96 ± 0.32 | 11.76 ± 0.13 | .492 | .109 |
| RBC, 1012/L | 11.27 ± 0.27 | 10.63 ± 0.22 | 10.66 ± 0.15 | 10.12 ± 0.16 | .061 | .067 |
| WBC, ×109/L | 18.87 ± 0.91 | 12.24 ± 1.20 | 13.67 ± 0.75 | 10.09 ± 0.91 | .0003 | .165 |
| PLT, ×109/L | 1173 ± 11.1 | 983 ± 33.3 | 1208 ± 11.7 | 929 ± 29.9 | .829 | .232 |
| MPV, fL | 7.3 ± 0.06 | 7.13 ± 0.04 | 7.18 ± 0.06 | 7.19 ± 0.06 | .299 | .309 |
| Pretamoxifen | End of study | Statistical significance, P | ||||
|---|---|---|---|---|---|---|
| AS/R3wt, n = 10 | AS/R3fl, n = 14 | AS/R3WT, n = 10 | AS/R3KD, n = 14 | AS/R3WT vs AS/R3wt | AS/R3KD vs AS/R3fl | |
| Hct, % | 36.74 ± 0.62 | 34.37 ± 0.69 | 35.19 ± 0.56 | 33.58 ± 0.58 | .079 | .389 |
| Hgb, g/dL | 13.08 ± 0.19 | 12.24 ± 0.21 | 12.74 ± 0.27 | 11.88 ± 0.16 | .323 | .188 |
| MCV, fL | 32.67 ± 0.5 | 32.3 ± 0.16 | 33.07 ± 0.68 | 33.16 ± 0.26 | .64 | .008 |
| RDW, % | 31.56 ± 1.23 | 29.99 ± 0.23 | 33.09 ± 1.84 | 31.51 ± 0.51 | .498 | .060 |
| MCH, pg | 11.66 ± 0.69 | 11.51 ± 0.07 | 11.96 ± 0.32 | 11.76 ± 0.13 | .492 | .109 |
| RBC, 1012/L | 11.27 ± 0.27 | 10.63 ± 0.22 | 10.66 ± 0.15 | 10.12 ± 0.16 | .061 | .067 |
| WBC, ×109/L | 18.87 ± 0.91 | 12.24 ± 1.20 | 13.67 ± 0.75 | 10.09 ± 0.91 | .0003 | .165 |
| PLT, ×109/L | 1173 ± 11.1 | 983 ± 33.3 | 1208 ± 11.7 | 929 ± 29.9 | .829 | .232 |
| MPV, fL | 7.3 ± 0.06 | 7.13 ± 0.04 | 7.18 ± 0.06 | 7.19 ± 0.06 | .299 | .309 |
AS/R3wt and AS/R3fl are AS/R3WT and AS/R3KD groups, respectively, before tamoxifen treatment. Values are mean ± SEM using the paired Student t test.
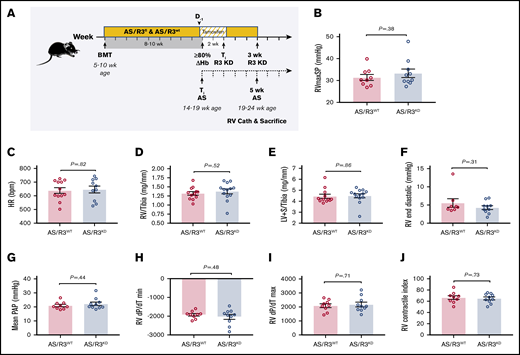
Three weeks of SMC CYB5R3 knockdown has no effect on cardiopulmonary hemodynamics or cardiac morphology in AS chimeras. (A) Study design for testing role of VSM CYB5R3 on hemodynamics and cardiac remodeling in AS chimeras. Cyb5r3fl/flMyh11-CreERT2 and Cyb5r3wt/wtMyh11-CreERT2 mice were lethally irradiated (500-550 rad twice) and transplanted with BM (2-4 million cells) from AS donor mice at 5 to 10 weeks of age to create AS/R3KD and AS/R3WT chimeras, respectively. At 8 to 10 weeks posttransplantation, peripheral blood was sampled to test engraftment to donor Hb phenotype. Chimeras testing 80% or more engraftment to AS phenotype, assessed by Hb electrophoresis, were fed a tamoxifen diet ad libitum for 2 weeks, and thereafter switched back to regular chow and aged for an additional 3 weeks before measurement of hemodynamics by RV microcatheterization. At 3 weeks CYB5R3 knockdown (solid line), animals were 5 weeks AS phenotype (dashed timeline) and a total age of 19 to 24 weeks. Complete blood counts were assessed at 8 to 10 weeks posttransplant, before and after tamoxifen diet (day −1 and day 14) and again at time of RV catheterization. No differences were noted between AS/R3KD (blue bar) and AS/R3WT (red bar) chimeras for RVmaxSP (n = 9-10) (B), heart rate (HR; n = 9-10) (C), RV weight normalized to tibia length (RV/tibia, n = 12-13) (D), LV+S weight normalized to tibia length (LV+S/tibia, n = 12-13) (E), RV end diastolic pressure (n = 9-10) (F), mPAP (n = 9-10) (G), RV dP/dT min (H), RV dP/dT max (n = 9-10) (I), and RV contractile index (n = 9-10) (J). The mean ± SEM is represented.
Three weeks of SMC CYB5R3 knockdown has no effect on cardiopulmonary hemodynamics or cardiac morphology in AS chimeras. (A) Study design for testing role of VSM CYB5R3 on hemodynamics and cardiac remodeling in AS chimeras. Cyb5r3fl/flMyh11-CreERT2 and Cyb5r3wt/wtMyh11-CreERT2 mice were lethally irradiated (500-550 rad twice) and transplanted with BM (2-4 million cells) from AS donor mice at 5 to 10 weeks of age to create AS/R3KD and AS/R3WT chimeras, respectively. At 8 to 10 weeks posttransplantation, peripheral blood was sampled to test engraftment to donor Hb phenotype. Chimeras testing 80% or more engraftment to AS phenotype, assessed by Hb electrophoresis, were fed a tamoxifen diet ad libitum for 2 weeks, and thereafter switched back to regular chow and aged for an additional 3 weeks before measurement of hemodynamics by RV microcatheterization. At 3 weeks CYB5R3 knockdown (solid line), animals were 5 weeks AS phenotype (dashed timeline) and a total age of 19 to 24 weeks. Complete blood counts were assessed at 8 to 10 weeks posttransplant, before and after tamoxifen diet (day −1 and day 14) and again at time of RV catheterization. No differences were noted between AS/R3KD (blue bar) and AS/R3WT (red bar) chimeras for RVmaxSP (n = 9-10) (B), heart rate (HR; n = 9-10) (C), RV weight normalized to tibia length (RV/tibia, n = 12-13) (D), LV+S weight normalized to tibia length (LV+S/tibia, n = 12-13) (E), RV end diastolic pressure (n = 9-10) (F), mPAP (n = 9-10) (G), RV dP/dT min (H), RV dP/dT max (n = 9-10) (I), and RV contractile index (n = 9-10) (J). The mean ± SEM is represented.
Effects of 12-week SMC Cyb5R3 knockdown on RVmaxSP and RV remodeling in SS chimeras
To determine the effects of longer SMC CYB5R3 knockdown on PH development in SS mice, similar to the preliminary study performed in C57BL/6J-transplanted chimeras (supplemental Figure 1), an additional study was conducted in which separate SS/R3KD and SS/R3WT groups were aged an additional 9 weeks (12 weeks total) following completion of tamoxifen treatment for SMC CYB5R3 knockdown (Figure 1). Knockdown of SMC-CYB5R3 for a 12-week period was chosen to establish CYB5R3 deficiency in SCD for a longer period and allow the chimeras enough aging with SCD to approximate the ages of the Berkeley SS mice that developed PH by 6 months of age in a prior study by Potoka et al.38 Closed-chest RV catheterization measurements of RVmaxSP in SS/R3KD (29.79 ± 1.09 mm Hg; n = 5) for 12 weeks were similar to the 3-week level (P = .32), whereas RVmaxSP in SS/R3WT at 12 weeks (31.18 ± 0.76 mm Hg; n = 6) had increased further (P = .005), essentially negating the intergroup difference that had been apparent at 3 weeks (Figure 2A,E). No intergroup difference in heart rate (Figure 2F) or other RV hemodynamic parameters (contractility, end diastolic pressure, dP/dT max, dP/dT min, and τ) (Figure 3G-L) were apparent between SS/R3KD and SS/R3WT. On the other hand, whereas the LV+S weight index was similar between SS/R3KD and SS/R3WT at 12 weeks (n = 5 and 6, respectively) (Figure 2H), the RV weight index for SS/R3KD remained enlarged relative to SS/R3WT (1.69 ± 0.06 vs 1.5 ± 0.06, respectively; n = 5-6 each) (Figure 2G), suggesting a more prolonged effect for SMC CYB5R3 deficiency on RV remodeling.
Relationship of SMC CYB5R3 protein expression with RVmaxSP
The quantity of CYB5R3 staining in SMC was determined via ImageJ using raw integrated intensity per medial SMC area, representing the CYB5R3 expression within the ACTA2+ area. Immunofluorescent staining of CYB5R3 protein in pulmonary artery sections from SS chimeras completing the 3- and 12-week knockdown studies (Figure 5A,D) showed roughly 60% reduction in SMC CYB5R3 protein in SS/R3KD (50.18 ± 9.70 and 41.87 ± 6.03, respectively) relative to SS/R3WT (124.5 ± 18.73 and 120.5 ± 9.05, respectively) (Figure 5B,E). The SMC-specific knockdown left CYB5R3 protein levels in endothelium unchanged (Figure 5C,F). Analysis of SMC CYB5R3 protein vs RVmaxSP showed a trending inverse relationship (r = −.52; P = .07) between the 2 parameters in the 3-week study (Figure 6A), linking increases in RVmaxSP to decreases in SMC CYB5R3. This finding suggests involvement of deficient SMC CYB5R3 expression and/or function in the early stages of SCD-associated PH development. On the other hand, there was no apparent relationship between SMC CYB5R3 protein and RVmaxSP in the 12-week study (r = .27; P = .46) (Figure 6D), implicating other molecular mechanisms, possibly compensatory, in the progression and/or continuance of PH at later time points.

Immunofluorescent staining of lung tissue from tamoxifen-treated SS chimeras shows knockdown of SMC CYB5R3 in pulmonary arteries of SS/R3KDanimals. Representative immunofluorescent images of postmortem lung tissue from SS/R3WT and SS/R3KD chimeras showing WT (left) and knockdown (right) levels of CYB5R3 at 3 weeks (A) and 12 weeks (D) following completion of tamoxifen-induced Cyb5r3 KO. A Nikon A1 confocal laser microscope was used to image the pulmonary arteries at ×40 magnification with 1096 × 1096 resolution. Z-stack imaging (1-µm increments) of stained and immunoglobulin G control sections were used for the maximum intensity projection representative images. Smooth muscle α actin (green), CYB5R3 (red), PECAM (gray), and DAPI-stained nuclei (blue). Quantification of Cyb5R3+ per ACTA2+ (smooth muscle) area in SS/R3WT (red bar) and SS/R3KD (blue bar) chimeras completing 3- (B) and 12-week (E) studies. Quantification of Cyb5R3+ per PECAM+ (endothelial) area in SS/R3WT and SS/R3KD chimeras completing 3- (n = 9-11) (C) and 12-week studies (n = 5 each) (F). Regions of interest were drawn onto the maximum intensity projection for ACTA2 (SMC), which was then superimposed on the maximum intensity projection for CYB5R3 using ImageJ software. The quantity of CYB5R3 staining in medial smooth muscle and endothelium was determined via ImageJ using raw integrated intensity per area. The mean ± SEM is represented. The Student unpaired t test was used to determine statistical significance.
Immunofluorescent staining of lung tissue from tamoxifen-treated SS chimeras shows knockdown of SMC CYB5R3 in pulmonary arteries of SS/R3KDanimals. Representative immunofluorescent images of postmortem lung tissue from SS/R3WT and SS/R3KD chimeras showing WT (left) and knockdown (right) levels of CYB5R3 at 3 weeks (A) and 12 weeks (D) following completion of tamoxifen-induced Cyb5r3 KO. A Nikon A1 confocal laser microscope was used to image the pulmonary arteries at ×40 magnification with 1096 × 1096 resolution. Z-stack imaging (1-µm increments) of stained and immunoglobulin G control sections were used for the maximum intensity projection representative images. Smooth muscle α actin (green), CYB5R3 (red), PECAM (gray), and DAPI-stained nuclei (blue). Quantification of Cyb5R3+ per ACTA2+ (smooth muscle) area in SS/R3WT (red bar) and SS/R3KD (blue bar) chimeras completing 3- (B) and 12-week (E) studies. Quantification of Cyb5R3+ per PECAM+ (endothelial) area in SS/R3WT and SS/R3KD chimeras completing 3- (n = 9-11) (C) and 12-week studies (n = 5 each) (F). Regions of interest were drawn onto the maximum intensity projection for ACTA2 (SMC), which was then superimposed on the maximum intensity projection for CYB5R3 using ImageJ software. The quantity of CYB5R3 staining in medial smooth muscle and endothelium was determined via ImageJ using raw integrated intensity per area. The mean ± SEM is represented. The Student unpaired t test was used to determine statistical significance.

Association of SMC CYB5R3 protein levels with RVmaxSP and biventricular cardiac remodeling in SS chimeras. CYB5R3 protein expression in pulmonary arterial SMC was determined for animals completing the 3- and 12-week studies. For the 3-week study, there was a trending association between SMC CYB5R3 protein expression and RVmaxSP: r = −.52, P = .069, n = 6-7 per group (A); corrected right ventricle mass (RV/Tibia): r = −.40, P = .10 (B); and corrected left ventricle + septum mass (LV+S/Tibia): r = −.43, P = .08; n = 8 per group for all (C). For the 12-week study, there was no obvious association of SMC CYB5R3 protein expression with RVmaxSP: r = .27, P = .46 (D); RV/Tibia: r = −.56, P = .09 (E); and LV+S/Tibia: r = −.17, P=.65; n = 5 per group for all (F). Correlation analyses were performed using Pearson r with significance set at <0.05.
Association of SMC CYB5R3 protein levels with RVmaxSP and biventricular cardiac remodeling in SS chimeras. CYB5R3 protein expression in pulmonary arterial SMC was determined for animals completing the 3- and 12-week studies. For the 3-week study, there was a trending association between SMC CYB5R3 protein expression and RVmaxSP: r = −.52, P = .069, n = 6-7 per group (A); corrected right ventricle mass (RV/Tibia): r = −.40, P = .10 (B); and corrected left ventricle + septum mass (LV+S/Tibia): r = −.43, P = .08; n = 8 per group for all (C). For the 12-week study, there was no obvious association of SMC CYB5R3 protein expression with RVmaxSP: r = .27, P = .46 (D); RV/Tibia: r = −.56, P = .09 (E); and LV+S/Tibia: r = −.17, P=.65; n = 5 per group for all (F). Correlation analyses were performed using Pearson r with significance set at <0.05.
Effects of SMC CYB5R3 knockdown on the intravascular blood cell compartment
The effect of SMC CYB5R3 knockdown on the intravascular blood cell phenotype in SS chimeras was determined by comparing hematology measurements made before starting tamoxifen treatment (day −1) and those made at the end of study (3- or 12-week knockdown) (Tables 1 and 3). Knockdown of SMC CYB5R3 for 3 weeks (Table 1) or 12 weeks (Table 3) produced no blood cell changes in SS chimeras.
Blood cell indices for Townes SS BM-transplanted Cyb5R3wt and Cyb5R3fl chimeras before tamoxifen treatment (day −1) and at end of 12-week study
| Pretamoxifen | End of study | Significance, P | ||||
|---|---|---|---|---|---|---|
| SS/R3wt, n = 7 | SS/R3fl, n = 5 | SS/R3WT, n = 7 | SS/R3KD, n = 5 | SS/R3WT vs SS/R3wt | SS/R3KD vs SS/R3fl | |
| Hct, % | 27.17 ± 2.22 | 24.08 ± 1.28 | 27.50 ± 1.65 | 22.98 ± 2.01 | .853 | .321 |
| Hgb, g/dL | 9.51 ± 0.72 | 8.62 ± 0.39 | 9.76 ± 0.56 | 8.24 ± 0.75 | .685 | .425 |
| MCV, fL | 48.4 ± 0.46 | 47.66 ± 0.67 | 45.59 ± 0.69 | 45.66 ± 0.75 | .005 | .075 |
| RDW, % | 35.17 ± 1.33 | 37.12 ± 1.05 | 37.8 ± 0.84 | 37.8 ± 2.43 | .014 | .822 |
| MCH, pg | 17.03 ± 0.26 | 17.1 ± 0.42 | 16.19 ± 0.24 | 16.4 ± 0.21 | .035 | .235 |
| RBC, 1012/L | 5.62 ± 0.46 | 5.07 ± 0.33 | 6.03 ± 0.34 | 5.05 ± 0.47 | .221 | .934 |
| WBC, ×109/L | 17.59 ± 2.46 | 12.64 ± 1.61 | 22.61 ± 2.46 | 11.46 ± 4.73 | .028 | .738 |
| PLT, ×109/L | 360 ± 49.6 | 301 ± 35.7 | 481 ± 44.7 | 448 ± 95.5 | .008 | .188 |
| MPV, fL | 6.76 ± 0.06 | 7.1 ± 1.64 | 6.7 ± 1.04 | 6.9 ± 0.12 | .76 | .286 |
| Pretamoxifen | End of study | Significance, P | ||||
|---|---|---|---|---|---|---|
| SS/R3wt, n = 7 | SS/R3fl, n = 5 | SS/R3WT, n = 7 | SS/R3KD, n = 5 | SS/R3WT vs SS/R3wt | SS/R3KD vs SS/R3fl | |
| Hct, % | 27.17 ± 2.22 | 24.08 ± 1.28 | 27.50 ± 1.65 | 22.98 ± 2.01 | .853 | .321 |
| Hgb, g/dL | 9.51 ± 0.72 | 8.62 ± 0.39 | 9.76 ± 0.56 | 8.24 ± 0.75 | .685 | .425 |
| MCV, fL | 48.4 ± 0.46 | 47.66 ± 0.67 | 45.59 ± 0.69 | 45.66 ± 0.75 | .005 | .075 |
| RDW, % | 35.17 ± 1.33 | 37.12 ± 1.05 | 37.8 ± 0.84 | 37.8 ± 2.43 | .014 | .822 |
| MCH, pg | 17.03 ± 0.26 | 17.1 ± 0.42 | 16.19 ± 0.24 | 16.4 ± 0.21 | .035 | .235 |
| RBC, 1012/L | 5.62 ± 0.46 | 5.07 ± 0.33 | 6.03 ± 0.34 | 5.05 ± 0.47 | .221 | .934 |
| WBC, ×109/L | 17.59 ± 2.46 | 12.64 ± 1.61 | 22.61 ± 2.46 | 11.46 ± 4.73 | .028 | .738 |
| PLT, ×109/L | 360 ± 49.6 | 301 ± 35.7 | 481 ± 44.7 | 448 ± 95.5 | .008 | .188 |
| MPV, fL | 6.76 ± 0.06 | 7.1 ± 1.64 | 6.7 ± 1.04 | 6.9 ± 0.12 | .76 | .286 |
SS/R3wt and SS/R3fl are SS/R3WT and SS/R3KD groups, respectively, before tamoxifen treatment. Values are mean ± SEM using the paired Student t test.
Plasma Hb concentrations at time of RV catheterization were similar between SS/R3KD and SS/R3WT chimeras completing the 3- and 12-week studies (supplemental Figure 4A-B). There was no significant correlation of plasma Hb level with any hemodynamic parameter in either SS/R3KD or SS/R3WT chimeras completing the 3-week study (supplemental Figure 5). Intergroup differences in correlations were also not apparent. These data indicate no difference between the SS/R3KD and SS/R3WT groups in vascular sensitivity to plasma Hb levels.
Effects of SMC CYB5R3 knockdown on myocardial fibrosis
The effects of SMC CYB5R3 knockdown on cardiac fibrosis was determined by comparing trichrome-stained right and left ventricles from SS/R3KD and SS/R3WT chimeras in the 3-week study, wherein intergroup differences in RVmaxSP were observed. Diffuse myocardial fibrosis was detected in both SS groups, which is in agreement with the recent findings of Bakeer et al,40 who reported enhanced cardiac magnetic resonance–detected extracellular volume in SS mice having both PH and LV diastolic dysfunction with preserved ejection fraction (heart failure with preserved ejection fraction [HFpEF]). No quantitative differences in cardiac fibrosis were observed between groups (supplemental Figure 6).
Discussion
Given the widespread organ pathology that results from living with SCD, even relatively modest elevations in PAP place individuals with SCD-associated PH at high risk for death from right-heart failure.12,21,41 Approximately one-half of these individuals have PH associated with high LV-filling pressures, categorized as group II, for which effective treatments are currently not available.16,42 Many SCD patients, whether they have PH or not, are nonresponsive to NO-based therapies. This suggests a defect in the NO-sGC-cGMP pathway that is downstream of NO bioavailability itself. It has been shown that CYB5R3 in aortic SMC is a key participant in the NO-sGC-cGMP pathway known to relax SMC and dilate vessels.28 sGC is vulnerable to oxidation, rendering its heme iron unable to bind NO and stimulate cGMP production. Through its reductase activity, CYB5R3 recycles oxidized heme (Fe3+) back to its reduced form (Fe2+), thereby maintaining NO-sGC-cGMP function. This is the first animal study to test SMC CYB5R3 involvement in SCD PH development. We show that spontaneous development of enlarged RVmaxSP and mPAP in a mouse model of SCD can be accelerated with diminished SMC CYB5R3 protein. The BM-transplanted SS chimeras used in this study were exposed to 5 or 15 weeks of circulating human Hb-S–containing erythrocytes, counting from the time ≥80% engraftment was detected to time of RV catheterization. When measured by RVmaxSP, the definitive measurement for diagnosing pulmonary arterial hypertension, PH has not been previously detected in transgenic SCD mice <3 months of age.7,43 Using BM transplant to confer SCD on phenotypically normal animals that were subsequently rendered deficient for SMC CYB5R3 using a tamoxifen-inducible KO approach, we reveal for the first time that SCD-associated PH can develop earlier than previously thought in SCD, namely by 5 weeks, when SMC CYB5R3 protein is deficient. Thus, this study shows that loss of CYB5R3 in SMC has an accelerating effect on the development of vascular and organ pathology that includes, but may not be limited to, the cardiopulmonary system in SCD.
Similar to other studies of PH in Townes or Berkeley SS mice,38,43,44 enlargement of both right and left ventricles was found in SS chimeras irrespective of SMC CYB5R3 expression. This study goes one step further and shows that 3 weeks of SMC CYB5R3 knockdown enhances biventricular hypertrophy in the SS chimeras. We recently showed in cultured rat aortic SMC that 1H-[1,2,4]oxadiazolo[4,3-a] quinoxalin-1-one–oxidized sGC generates substantially decreased NO-stimulated cGMP when CYB5R3 protein is absent.28 Induction of SMC CYB5R3 knockdown in SS mice, wherein oxidative stress exists, mimics this situation under in vivo conditions. Hence, it is reasonable to attribute the elevated RVmaxSP and RV hypertrophy in SS chimeras with 3 weeks knockdown of SMC CYB5R3 to impairment of sGC’s ability to sense NO, relax SMC, and lower pulmonary arterial resistance. There are likely widespread effects of SMC CYB5R3 knockdown on sGC regulation of vascular tone, which manifest as increased PVR. Although PVR was not measured, the LV hypertrophy in SS chimeras with 3 weeks SMC CYB5R3 knockdown strongly support this interpretation. To the extent that LV remodeling may reflect impairments to LV function, one questions how much PH development in SCD originates on the left side of the heart due to high postcapillary or venous pressure in the lungs. Indeed, LV diastolic dysfunction commonly accompanies HFpEF in patients with SCD-associated PAH (group 2 PH).13,42,45,46 The technical challenges of working with SS mice prevented us from reliably measuring LV hemodynamics using open-chest microcatheterization. When considering other possible explanations for ventricular remodeling, one could invoke the chronic anemia of SCD as a contributing factor in RV hypertrophy. Anemia-driven increases in heart rate can adversely affect RV afterload, with increased work for the RV spurring compensatory remodeling.47 The augmented RV end diastolic pressure observed in SS chimeras with 3 weeks of SMC CYB5R3 knockdown is consistent with an elevated RV preload that one expects with anemia.48,49 Although the monogenetic defect of sickle cell anemia, resulting in substitution of valine for glutamic acid in the codon for β-globin synthesis, is certainly the origin of the anemia that drives much of the multiorgan pathology known to characterize the disease, the degree of proximity with which that anemia affects ventricular remodeling or function on either side of the heart remains unclear. Our findings of similar levels of anemia and heart rates between the CYB5R3 knockdown and WT chimeric animals in the 3-week study cannot be used to support a role for anemia-driven effects on the disparate RV remodeling. There were changes to mean corpuscular volume (MCV) and mean corpuscular Hb (MCH) over time (3 weeks and 12 weeks after completion of tamoxifen treatment), but these changes were only noted in the SS animals with WT Cyb5r3 and not in their knockout (KO) counterparts. Thus, we conclude that the acceleration of PH development in the SS knockdown animals resulted specifically from the CYB5R3 deficit in the vascular SMC compartment.
Red blood cell hemolysis contributes to anemia of SCD and generates cell-free plasma Hb products that can be elevated in the absence of sufficient Hb and heme-scavenging mechanisms (haptoglobin and hemopexin, respectively), which have been shown to be overwhelmed in patients and animal models with the disease.50,51 We did not observe a significant difference in plasma Hb levels between the CYB5R3 knockdown and WT groups at either the 3- or 12-week time points. The absence of differences in blood cell counts between the CYB5R3 knockdown and WT groups is consistent with these findings. Thus, these data do not support a role for enhanced scavenging of NO or generation of reactive oxygen species by cell-free Hb or heme in the accelerated PH development observed in the 3-week CYB5R3 knockdown group. However, it is conceivable that the deficiency of CYB5R3 in vascular SMC obstructs reductive recycling of oxidized sGC,28 enabling a constricted vascular environment relative to WT animals.38 In such an environment even small, statistically insignificant increases in cell-free Hb and heme may be sufficient to accelerate PH development.4,10,41 The sum of our hemodynamic and hematological findings is more consistent with impaired sGC-mediated pulmonary arteriolar dilation in CYB5R3-deficient vascular SMC (Figure 7).
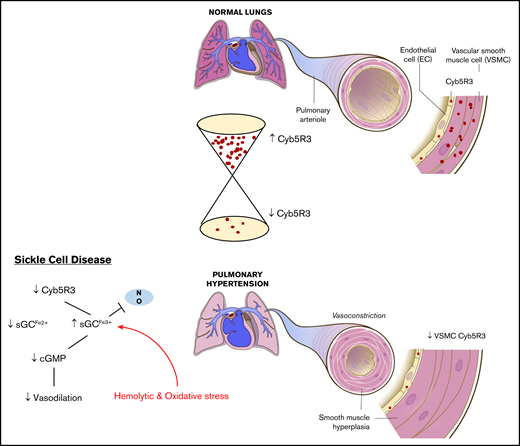
Schematic of CYB5R3 protein expression in pulmonary arterial SMCs and its effect on pulmonary artery vascular tone. Under normal conditions CYB5R3 in VSM regulates the redox state of sGC heme iron, maintaining sGC in its NO-sensitive reduced form (Fe2+), whereby signaling for cGMP production occurs and mediates vessel relaxation for low pulmonary vascular resistance. In SCD, CYB5R3 protein expression in VSM is relatively low and sGC heme iron oxidation (Fe3+) is high due to chronic hemolytic and oxidative stress. The result is “resistance” to NO signaling for cGMP production. This pathology produces a constricted pulmonary arterial phenotype, in which high vascular resistance increases pressure to drive pulmonary arterial hypertension and hyperplasia.
Schematic of CYB5R3 protein expression in pulmonary arterial SMCs and its effect on pulmonary artery vascular tone. Under normal conditions CYB5R3 in VSM regulates the redox state of sGC heme iron, maintaining sGC in its NO-sensitive reduced form (Fe2+), whereby signaling for cGMP production occurs and mediates vessel relaxation for low pulmonary vascular resistance. In SCD, CYB5R3 protein expression in VSM is relatively low and sGC heme iron oxidation (Fe3+) is high due to chronic hemolytic and oxidative stress. The result is “resistance” to NO signaling for cGMP production. This pathology produces a constricted pulmonary arterial phenotype, in which high vascular resistance increases pressure to drive pulmonary arterial hypertension and hyperplasia.
It is important to note that the enhanced RV remodeling in SS animals with SMC CYB5R3 knockdown occurred without apparent changes to RV contractility. This finding suggests that RV failure requires more time to evolve, which is consistent with a report by Hsu et al that 3- to 5-month-old Berkeley SS mice with PH could still increase cardiac output in response to anemia.7 Even though RV failure may only appear in older SS mice,7 RV remodeling can still negatively impact the LV. RV end diastolic pressure, reflective of RV preload, was increased in SS chimeras with 3 weeks of SMC Cyb5R3 knockdown. Elevated RV preload can result in RV encroachment on space occupied by the interventricular septum, decreasing LV compliance and performance even in the absence of overt LV failure.52 Hence, elements of cardiac remodeling, which predispose to cardiac failure, can already exist at 5 weeks of life with SCD, as indicated by the biventricular myocardial fibrosis detected in both SS groups completing the 3-week study.
Whereas level of SMC CYB5R3 protein seems critical to NO-sGC-cGMP–mediated vascular relaxation, our inability to detect any SMC sGC protein in pulmonary arteries of SS/R3KD and SS/R3WT chimeras in either the 3- or 12-week studies (data not shown) suggests that even low sGC protein levels can mediate vasorelaxation. Immunofluorescent staining for sGC protein was detected in SMC of AS/R3WT pulmonary arteries and in SMC of C57BL/6J aortas. The likelihood of sGC being oxidized or heme-deficient in SCD is great.53 Drugs that boost sGC’s function by (1) activation of its oxidized or heme-deficient form (cinaciguat) or (2) stimulation of its NO-dependent reduced form (riociguat) can be used to indirectly interrogate sGC protein expression that is below the level of detection by currently available antibodies. These drugs would also help determine the extent to which Cyb5r3 KO effects are sGC-dependent. This is a future direction for our group.
Even though improved supportive care has extended the lifespans of individuals with SCD, cardiac and pulmonary complications contribute to most deaths in SCD patients.54 Analysis of CYB5R3 expression in this population may predict risk for early PH development and opportunity for early intervention. Over 40 naturally occurring polymorphisms of CYB5R3 have been identified in the human population.55 The rs1800457 variant of CYB5R3 has decreased function due to a key serine-to-threonine substitution. With this variant being found only in persons of African ancestry with a high allelic frequency of 0.23,56 it is imperative that future studies investigate how this CYB5R3 variant participates in SCD pathology and determines interindividual responses to therapy. Patients with more PH, in terms of severity or age of onset, may be carriers of loss-of-function CYB5R3 variants.
Acknowledgments
The authors thank the University of Pittsburgh Vascular Medicine Institute Phenotyping Core for technical assistance with acquisition of hemodynamic data. The authors also thank Michele Mulkeen of the Rangos Research Center at the University of Pittsburgh Medical Center Children’s Hospital of Pittsburgh for assistance with histology.
This work was supported, in whole or in part, by National Institutes of Health grants: National Heart, Lung, and Blood Institute grant R25 HL128640-03 (K.C.W.); National Institute of Diabetes and Digestive and Kidney Diseases grant T32 DK00705244 (B.G.D.); National Heart, Lung, and Blood Institute grant K01 HL133331 (D.A.V.); National Heart, Lung, and Blood Institute grants R01 HL106192 and U01HL117721 (S.F.O.-A.); National Heart, Lung, and Blood Institute grant P01 HL103455-06 (A.L.M.); National Heart, Lung, and Blood Institute grants R01 HL098032, R01 HL125886-01, P01HL103455, and T32 HL007563 (M.T.G.); and National Heart, Lung, and Blood Institute grants R01 HL133864 and R01 HL128304, and an American Heart Association Established Investigator Award (A.C.S.). This work was also supported by the Institute for Transfusion Medicine and the Hemophilia Center of Western Pennsylvania (K.C.W., S.F.O.-A., A.L.M., M.T.G., and A.C.S.).
Authorship
Contribution: K.C.W., A.C.S., J.J.B., S.A.H., B.G.D., H.M.S., and D.A.V. conceived, designed, and performed the experiments; S.A.H., S.F.O.-A., and S.G. maintained animal colonies; K.C.W., A.C.S., B.G.D., H.M.S., T.B., A.L.M., and M.T.G. analyzed and interpreted data; K.C.W., A.C.S., and M.T.G. drafted and revised the manuscript; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: A.C.S. receives research support from Bayer Pharmaceuticals. M.T.G. is listed as a coinventor on a National Institutes of Health government patent for the use of nitrite salts in cardiovascular diseases (licensed by United Therapeutics) and on provisional patents for the use of recombinant neuroglobin and heme-based molecules as antidotes for CO poisoning (licensed by Globin Solutions, Inc), and is a coinvestigator in a research collaboration with Bayer Pharmaceuticals to evaluate riociguat as a treatment for patients with SCD. D.A.V. is an ad hoc consultant for Complexa Inc. The remaining authors declare no competing financial interests.
Correspondence: Adam C. Straub, Heart, Lung, Blood and Vascular Medicine Institute, Department of Pharmacology and Chemical Biology, School of Medicine, University of Pittsburgh, E1254 Biomedical Science Tower, 200 Lothrop St, Pittsburgh, PA 15216; e-mail: astraub@pitt.edu.

